Distrofia muscular de Duchenne
A distrofia muscular de Duchenne (DMD) é uma doença neuromuscular genética rara causada por mutações no gene da distrofina.7 Caracteriza-se pela deterioração progressiva da função muscular, levando à perda da marcha e à insuficiência respiratória e cardíaca.7 A DMD pertence ao grupo das distrofinopatias, um grupo de doenças neuromusculares causadas por alterações no gene da distrofina (DMD).
As doenças neuromusculares abrangem uma vasta gama de doenças que afetam os músculos e os nervos que os controlam.1
São geralmente classificados de acordo com a localização da doença e incluem:1
- Perturbações musculares (por exemplo, distrofia muscular de Duchenne)
- Perturbações da junção neuromuscular (por exemplo, síndrome miasténica congénita)
- Perturbações dos neurónios motores (por exemplo, atrofia muscular espinal)
- Perturbações nervosas periféricas (por exemplo, doença de Charcot-Marie-Tooth)
A maioria das doenças neuromusculares que ocorrem na infância apresentam uma base genética.1 A doença neuromuscular genética pediátrica mais comum é a distrofia muscular de Duchenne, que afeta 1 em 3.600 a 6.000 recém-nascidos em todo o mundo.2,3
Quais são os sinais e sintomas das perturbações neuromusculares?
Crianças com uma doença neuromuscular sofrem uma deterioração progressiva dos músculos e células musculares e um declínio contínuo da sua função física.4 O primeiro e mais comum sinal de doença neuromuscular é a fraqueza muscular progressiva, que se manifesta como um atraso no desenvolvimento motor.5,6 Os atrasos na linguagem, na fala e na cognição também devem levantar suspeitas de que o doente possa ter uma desordem neuromuscular.6
A monitorização do desenvolvimento motor pode ajudar a identificar atempadamente quaisquer atrasos no desenvolvimento, permitindo o rápido encaminhamento para um especialista e assim ajudar no processo de diagnóstico precoce.5,7
Porque é importante um diagnóstico precoce?
Embora as doenças neuromusculares não tenham atualmente uma cura, existem opções de monitorização e tratamento disponíveis que melhoram a progressão da doença.4
A distrofia muscular de Duchenne (DMD) é uma doença neuromuscular genética rara causada por mutações no gene da distrofina.7 Caracteriza-se pela deterioração progressiva da função muscular, levando à perda da capacidade de marcha e à insuficiência respiratória e cardíaca.7
A DMD é um tipo de distrofia que pertence ao grupo das distrofinopatias, um grupo de doenças musculares causadas por mutações no gene da distrofina para o qual não existe cura. Os sintomas podem variar de ligeiros (hiperCKaemia assintomática) a graves e progressivos (distrofia muscular de Becker [DMB] e DMD).8-11
O que é a distrofina?
A distrofina ajuda a manter a estrutura das fibras musculares e proporciona estabilidade da membrana durante a contração.12 É expressa no músculo esquelético e cardíaco e está situada na face citoplasmática da membrana da fibra muscular ou sarcolema.13-16 Como parte do complexo distrofina-glicoproteína, a distrofina liga o aparelho contráctil intracelular à matriz extracelular dos miócitos, que fornece suporte mecânico às membranas do músculo esquelético ou cardíaco durante a contração.12-16
A DMD é causada por mutações no gene da distrofina que reduzem ou inibem a produção da proteína da distrofina.7,8
O gene da distrofina é um dos maiores genes humanos conhecidos e contém 79 exões.17
No total, foram identificadas mais de 7.000 mutações no gene da distrofina, incluindo supressões, duplicações e pequenas mutações, muitas das quais são de importância clínica.17,18
A DMD é uma doença recessiva ligada ao X que afeta principalmente os homens.3,19
As mutações no gene da distrofina mostram um padrão recessivo ligado ao X. Quando a mutação é herdada, há 50% de probabilidade de que a descendência masculina de uma mãe afetada possa herdar o cromossoma defeituoso15. As filhas são igualmente susceptíveis de herdar o cromossoma defeituoso, mas a mutação é compensada pela presença de uma cópia normal da distrofina herdada do pai, o que frequentemente resulta em formas assintomáticas ou mais suaves da doença nas raparigas.3,20
Existem diferentes tipos de mutações que inibem a produção da distrofina proteica funcional total.21
As mutações mais frequentes observadas no gene da distrofina são17,21:
Em geral, os fenótipos mais graves de DMD estão associados a mutações que alteram o quadro de leitura e resultam numa proteína truncada e não funcional. A distrofia muscular de Becker (DMB) é uma doença hereditária e progressiva que causa uma deficiência muscular mais ligeira em comparação com a DMB. Tem um fenótipo mais variável e, na maioria dos casos, está associada a supressões não estruturais que resultam numa proteína distrofina anormal, mais pequena, mas funcional10,20,21. A hipótese do quadro de leitura explica mais de 90% dos casos, no entanto, existem exceções20. Este é o caso de doentes com DMB causada por deleções ou duplicações com mudança de quadro de leitura ou DMB com deleções ou duplicações sem mudança de quadro de deleção.20
A deficiência ou ausência de distrofina causa fraqueza muscular e fibrose.22,23
A deficiência ou ausência de distrofina provoca a perda da ligação do citosqueleto actínico ao tecido conjuntivo e danifica as fibras musculares durante a contração, resultando em dor muscular crónica, inflamação e, por fim, as fibras musculares são substituídas por gordura e tecido fibrótico com subsequente perda da função muscular.22
Processo de degeneração e perda de massa muscular como consequência da ausência de distrofina.22,23
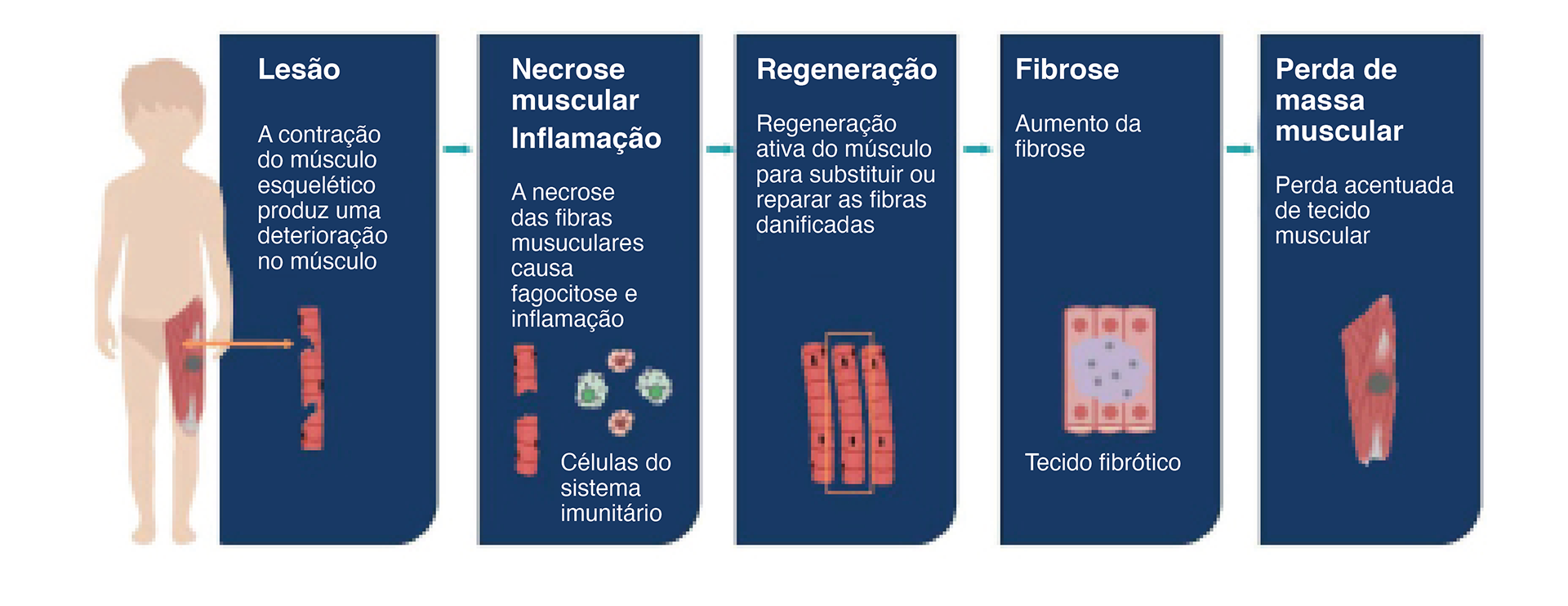
A perda de tecido muscular tem início na infância e é irreversível.3,24
A ausência de distrofina em doentes com DMD resulta em desgaste contínuo dos músculos e na substituição das fibras musculares por tecido cicatrizado e gordura.23,24
Com o tempo, o desgaste muscular é contínuo e as fibras musculares são substituídas por tecido cicatrizado e gordura25
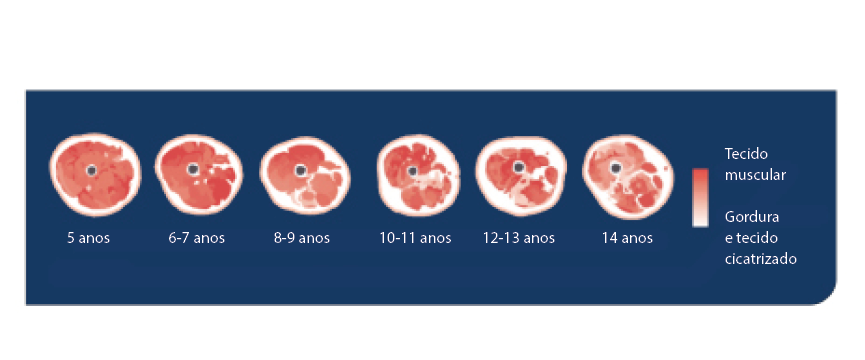
As imagens mostram infiltração de gordura e tecido fibrótico no músculo vastus lateralis (a idade em anos).
Figura adaptada de Sweeney HL.25
. Aos 5 anos de idade, a fraqueza muscular é evidente e observa-se uma redução de 50-60% na força muscular.26
. Aos 6 anos de idade, apenas 60% da massa muscular esperada é retida, que é reduzida para 20% aos 16 anos
de idade.27
A prevalência da DMD é de 1 em 3.600 a 6.000 nascimentos do sexo masculino, aproximadamente 30% dos casos não têm antecedentes familiares e apenas 10% das mulheres portadoras apresentam quaisquer sintomas da doença.3,28-30
Embora o fenotipo das raparigas afetadas seja geralmente muito mais ligeiro do que o dos rapazes, em alguns casos raros de rearranjo cromossómico ou inativação do cromossoma X, a gravidade da doença torna-se semelhante.3
Nos Estados Unidos, a prevalência estimada da distrofia muscular de Duchenne e de Becker em 2010 foi de 1,38 por 10.000 homens, de acordo com dados e estatísticas da Muscular Dystrophy Surveillance, Tracking and Research Network (MD STARnet).31-32 A prevalência da DMD era três vezes mais elevada do que a da DMB.31
A dificuldade em reconhecer os sinais e sintomas da DMD pode atrasar o diagnóstico.5,6
Uma das principais dificuldades para o diagnóstico inicial é a mutação espontânea do gene DMD. Isto ocorre em cerca de um terço dos casos em que ocorrem mutações espontâneas sem um historial familiar de DMD.23
Em doentes com DMD:
. O atraso médio no diagnóstico (desde o início dos primeiros sintomas) é entre 1,3 e ~2,5 anos.28,34
. A idade média no diagnóstico situa-se entre 4,3 e 4,5 anos.6,34
O diagnóstico de DMD pode ser atrasado por várias razões. Em primeiro lugar, a heterogeneidade de apresentação pode tornar difícil a identificação pelos profissionais de saúde. Outra razão é a relutância dos pais em procurar um diagnóstico devido a sentimentos contraditórios, como se tem reflectido em alguns inquéritos realizados no passado.35
O reconhecimento dos sinais e sintomas indicativos da doença
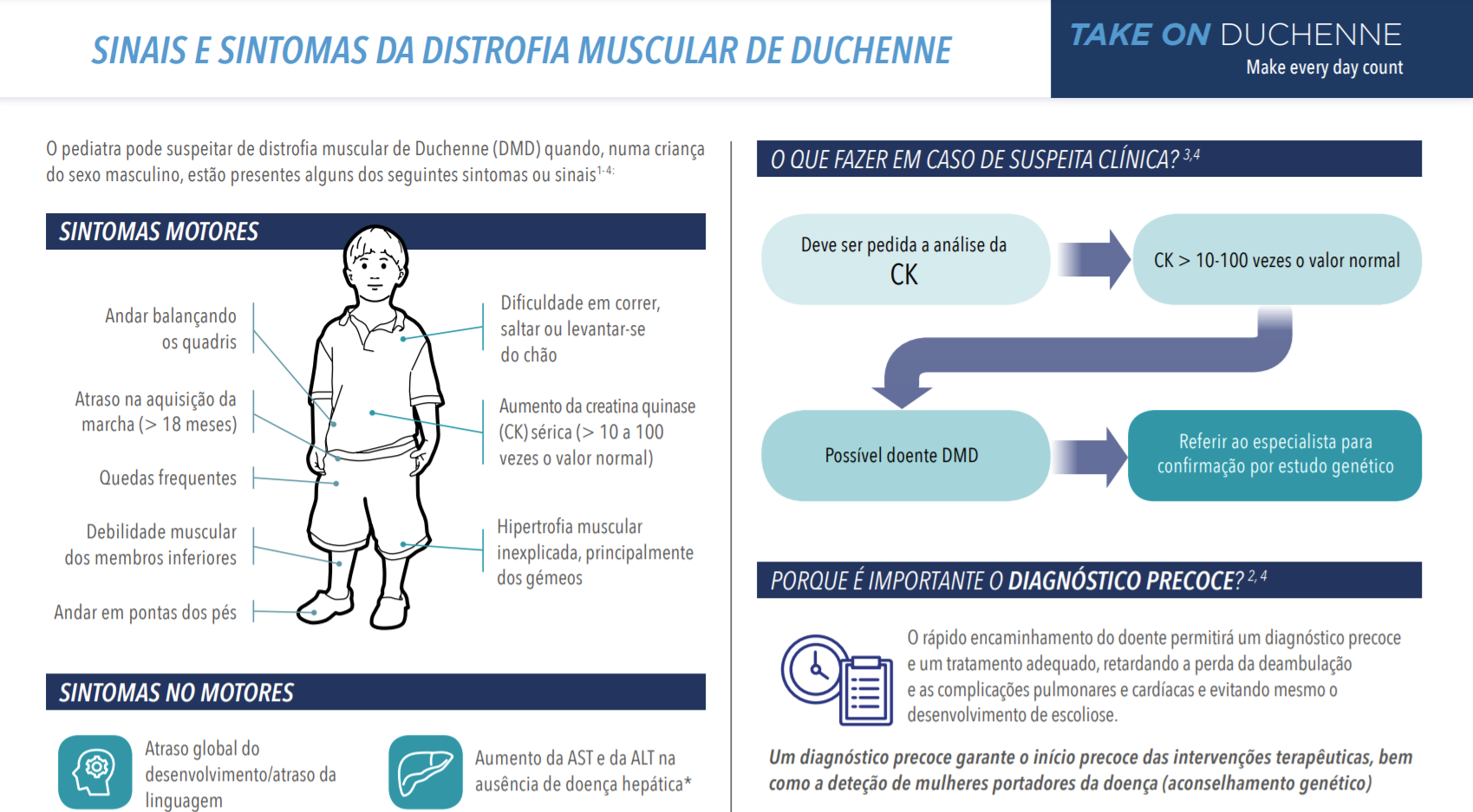
A suspeita de DMD pode surgir na primeira infância devido a atrasos na realização de marcos motores, tais como o sentar e ficar de pé de forma independente. Os primeiros sintomas de DMD identificados pelos pais são frequentemente atrasos gerais no desenvolvimento motor, tais como falta de jeito e quedas frequentes, problemas relacionados com a marcha, por exemplo, ponta dos pés, pés chatos e atraso no início da marcha. Outros sintomas que podem levantar suspeitas de DMD são problemas relacionados com a aprendizagem e a fala.7,8
O sinal de Gowers, uma manobra usada para se levantar da posição supina em que a criança usa os membros superiores e se inclina sobre as pernas para compensar a fraqueza muscular pélvica, é também um sinal precoce frequentemente observado nesta doença.36,37
Manobra de Gowers38
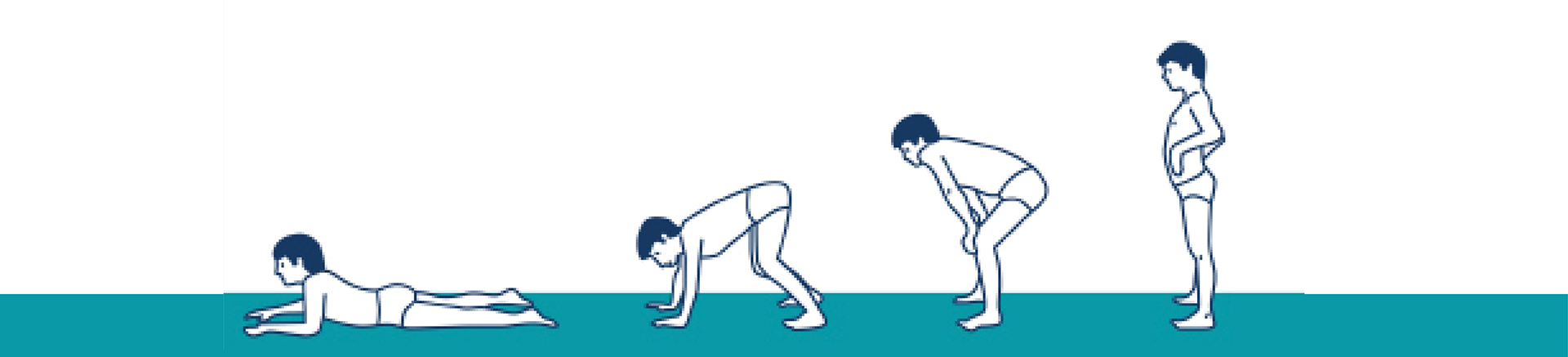
A identificação precoce de atrasos no desenvolvimento motor permite o encaminhamento atempado para intervenção, exames de diagnóstico e planeamento do tratamento.39
Sintomas motores7,28,30,37


Outros sinais e sintomas de DMD:
- Elevação da creatina quinase sérica (CK) ou transaminases.7,40-42
- Atraso cognitivo.7,30
- Hipertrofia dos músculos da barriga das pernas.7,30
- Marcha anormal.7,28
Sintomas não motores



TDAH

* Em fases pré-sintomáticas da doença, a elevação de aspartato aminotransferase, enzimas produzidas tanto por células musculares como hepáticas, pode levar erroneamente à suspeita de doença hepática.
Uma concentração elevada de CK é um sinal de lesão muscular e é indicativa da presença de algumas doenças neuromusculares.44,46 Deve ser realizado um exame à CK quando:
dificuldade em estar de pé ou incapacidade de caminhar aos 16-18 meses.5,7
Determinar o diagnóstico de DMD
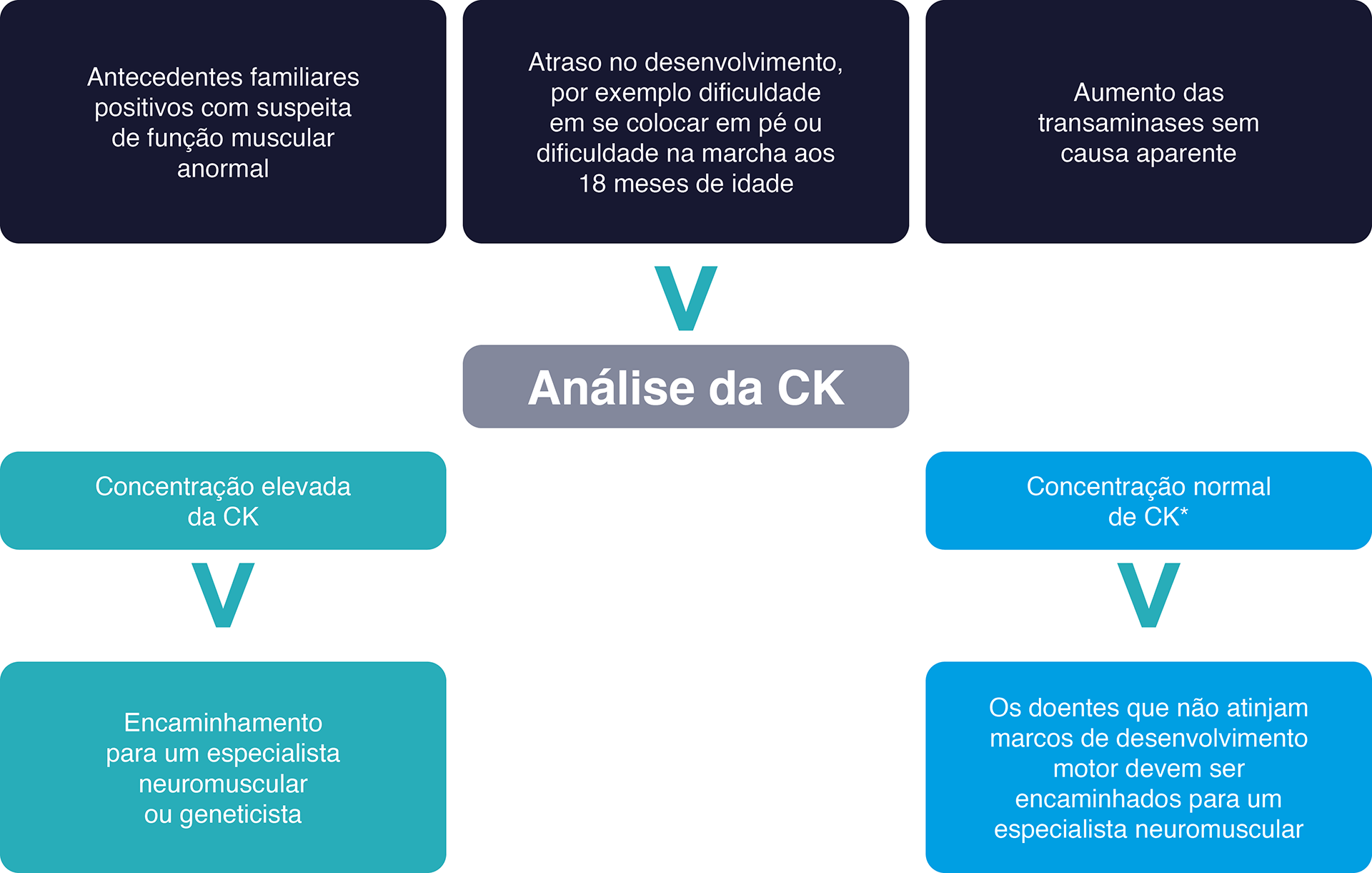
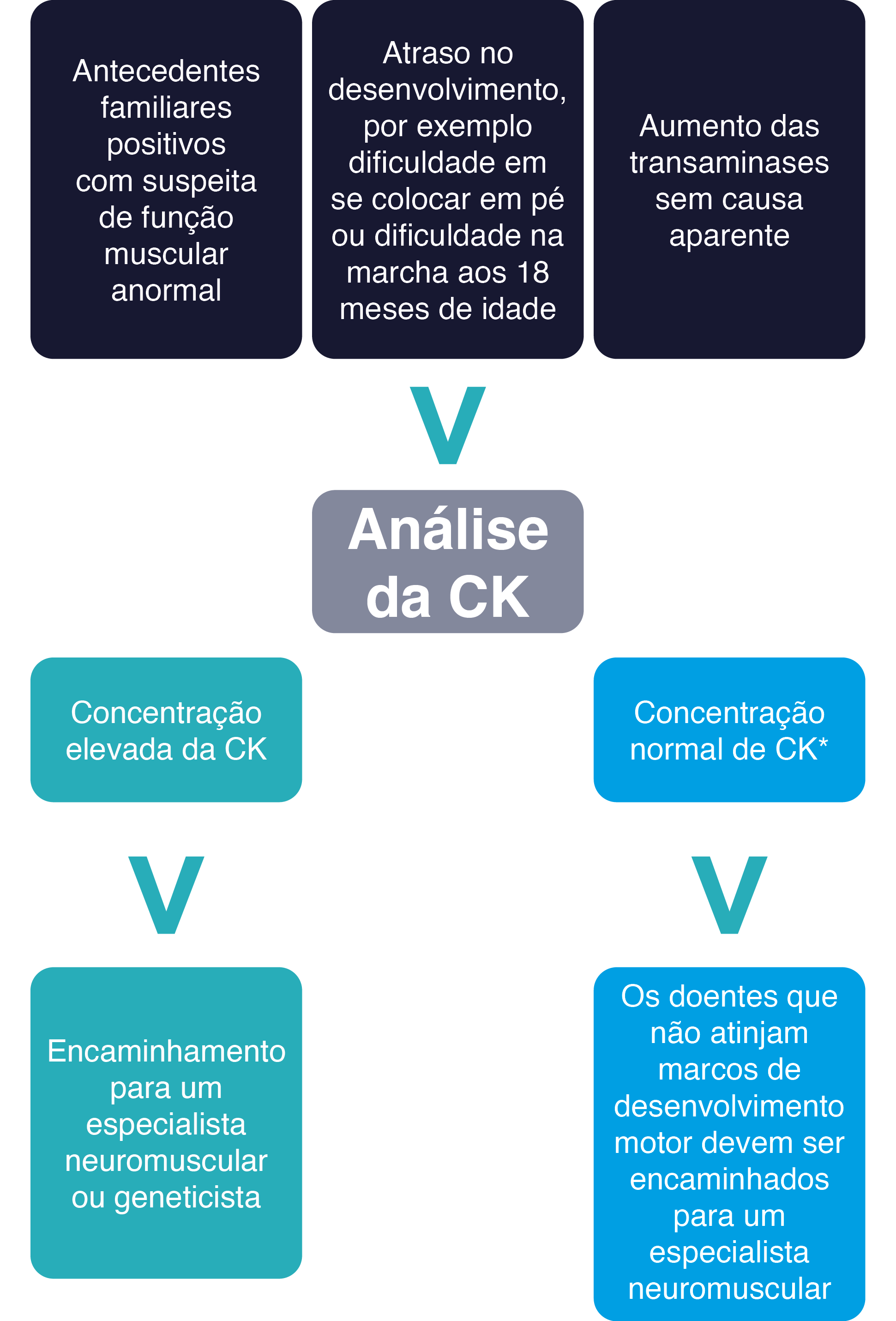
CK, creatina quinase;L, litros; U, unidades.
* A gama normal de CK é normalmente de até 250 U/L. Os valores absolutos podem diferir de laboratório para laboratório.
Os exames genéticos podem confirmar a presença de DMD.
Para confirmar a presença de DMD, é necessário realizar um diagnóstico genético e identificar a mutação específica causadora da doença.7 Este método é o único que pode determinar a mutação específica do doente.7,21
- Conhecer a mutação específica causadora de DMD é importante porque permite determinar as melhores opções
de tratamento, bem como a possibilidade de participar em ensaios clínicos.21
- Os exames de portadoras podem ajudar a reduzir a transmissão de DMD e a melhorar os resultados para
as mulheres em risco.7,48
Como conseguir um diagnóstico genético preciso da DMD?7,30,44
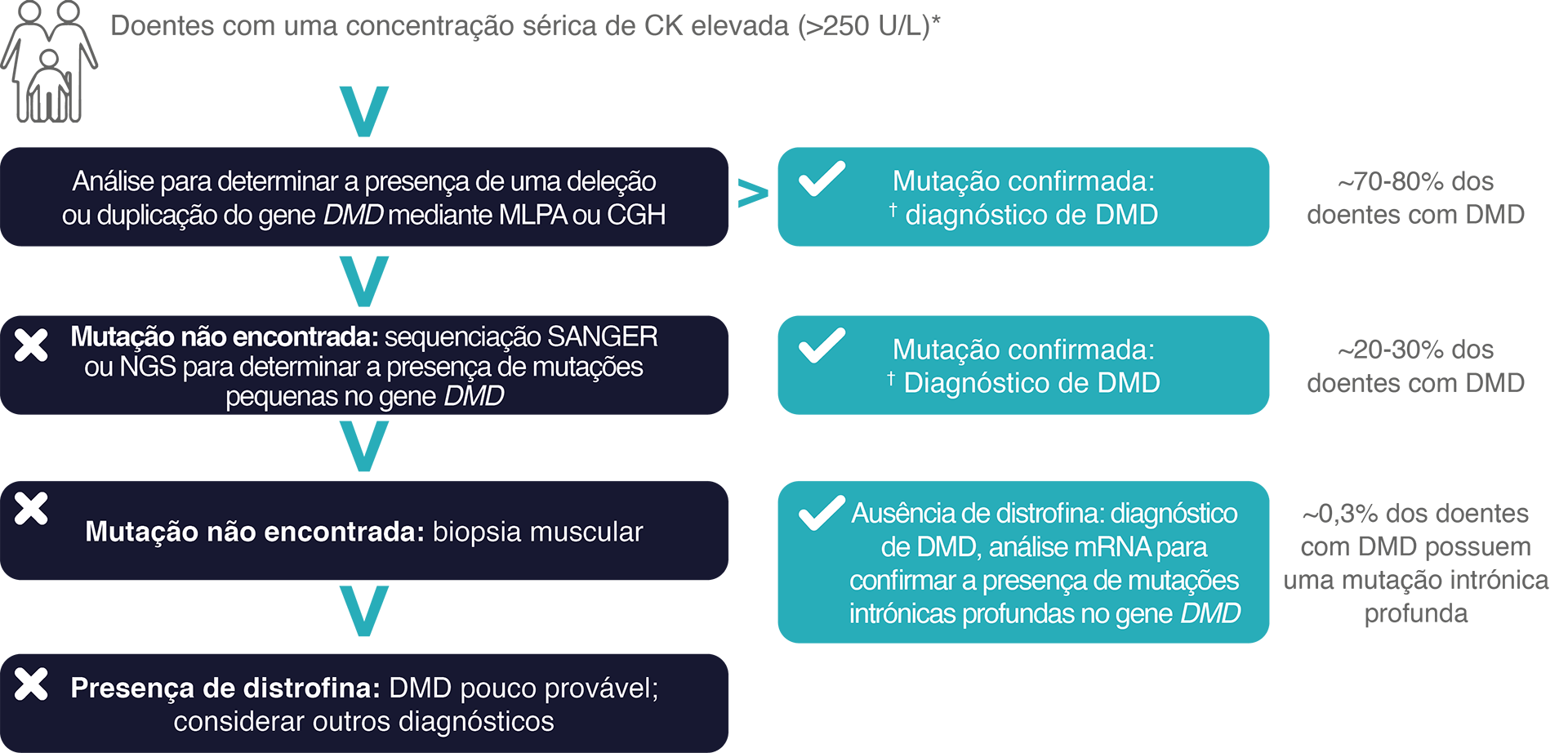
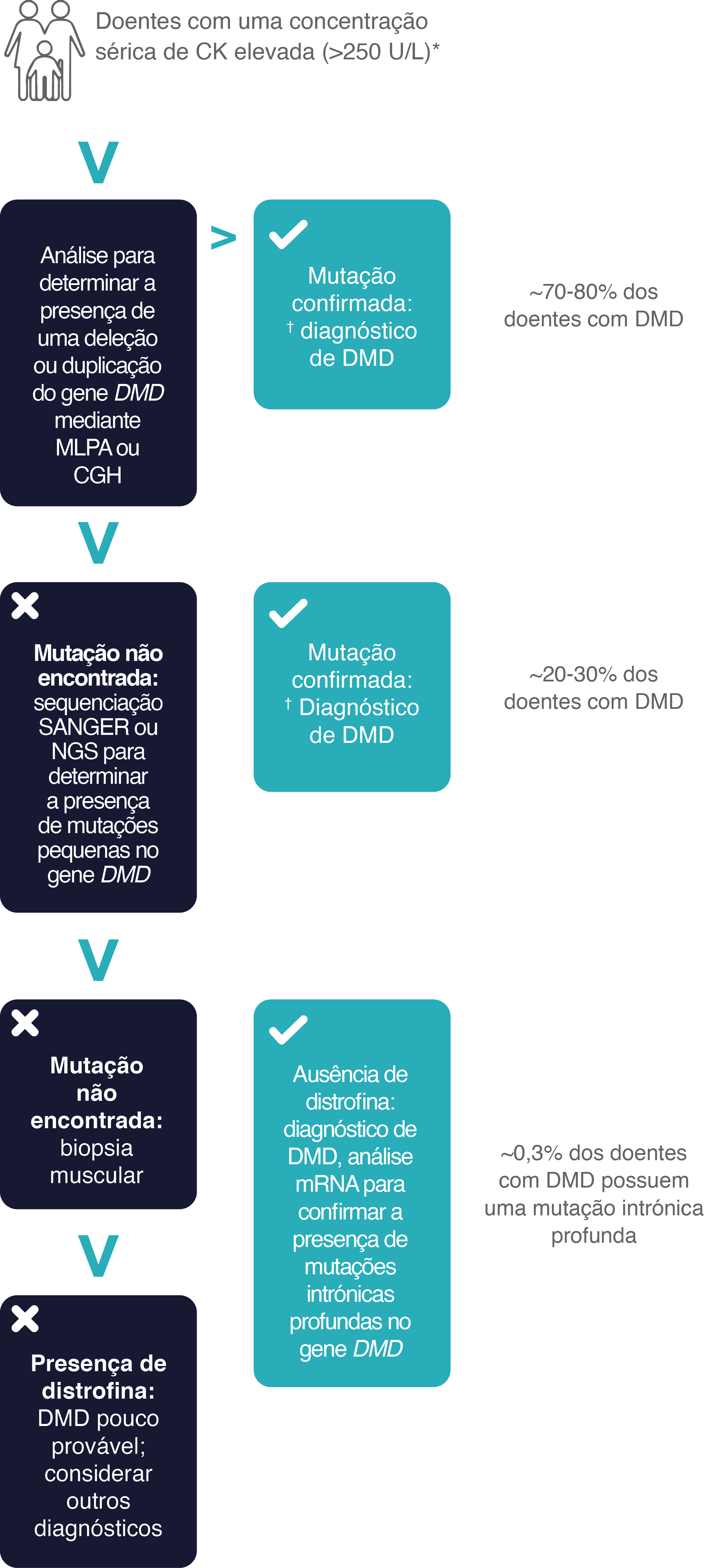
CGH, hibridação genómica comparativa; CK, creatina quinase; L, litros; MLPA,
amplificação de sonda dependente de ligação multiplex; mRNA, RNA mensageiro; NGS, sequenciação de última geração; U, unidades.
* O intervalo normal de CK é normalmente de até 250 U/L. Os valores absolutos podem diferir de laboratório para laboratório.30
† Regra do marco de leitura: Em geral, as mutações in-frame de leitura confirmam a presença de DMD e as mutações out-of-frame de leitura confirmam
a presença de DMB, uma forma menos grave da doença. Aproximadamente 4,9% das mutações no gene DMD não seguem a regra do marco de leitura.30
A história natural da DMD segue uma evolução devastadora e progressiva durante a qual os pacientes perdem a capacidade funcional.7
Progressão da doença em doentes com DMD6,26,49
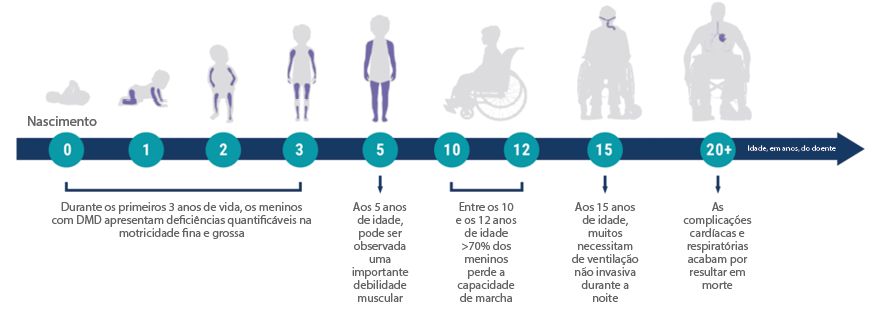
As intervenções que podem modificar a história natural da DMD são mais benéficas quanto menos danificado estiver o músculo e quanto mais jovens forem os doentes.6,15,34
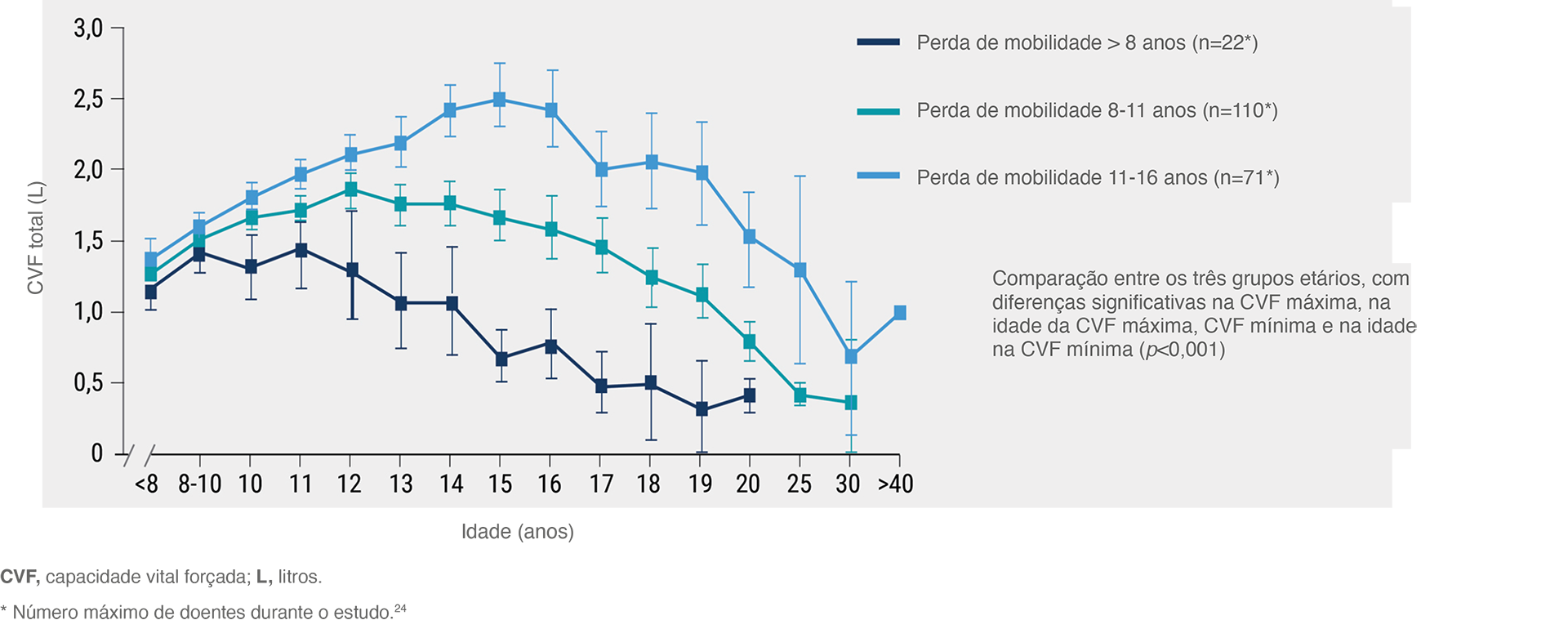
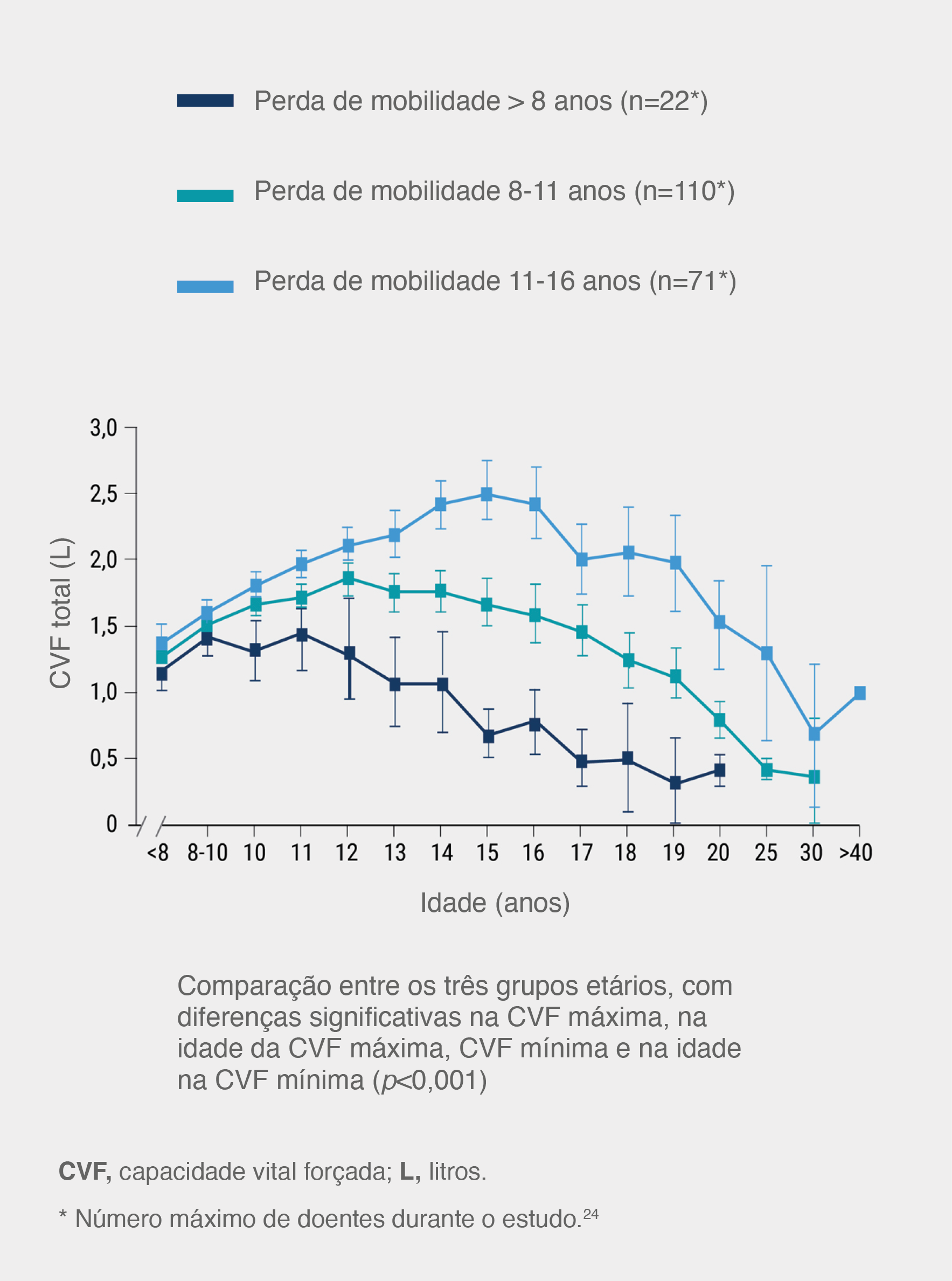
A perda de mobilidade é um fator-chave na previsão da progressão da doença.24
A perda da capacidade de andar é um preditor chave da progressão da DMD, que eventualmente leva a graves problemas respiratórios e à morte por insuficiência respiratória.24 A perda da capacidade de caminhar numa idade mais precoce está associada a uma insuficiência respiratória mais precoce e mais grave.24
Função física durante a etapa com mobilidade na DMD: comparação de três grupos etários24
O tratamento da DMD envolve muitos profissionais de saúde e a coordenação entre eles é essencial para o máximo benefício terapêutico.7
As orientações internacionais de cuidados na DMD recomendam que, uma vez confirmado o diagnóstico, um especialista neuromuscular deve coordenar o trabalho dos diferentes especialistas e intervenções7. A melhoria da sobrevivência deve-se às várias subespecialidades que cuidam dos doentes de Duchenne, utilizando estratégias mais proativas que favorecem a prevenção, o diagnóstico precoce e um melhor tratamento das complicações previsíveis e potencialmente modificáveis da doença.7
De acordo com as recomendações publicadas pelo grupo internacional de peritos em cuidados de DMD, os doentes, prestadores de cuidados e profissionais de saúde devem coordenar-se para cumprir os objetivos de tratamento a longo prazo e utilizar os recursos disponíveis para um planeamento específico, tendo em conta a natureza da DMD.50
Os doentes com DMD requerem múltiplas adaptações para a sua vida diária e a acessibilidade do meio ambiente torna-se um problema quando perdem a função dos membros inferiores, membros superiores e capacidade respiratória.50,51
As adaptações para a vida diária são necessárias desde as fases iniciais da DMD, por exemplo:7,50
É necessária uma transição de cuidados da adolescência para a idade adulta para ajudar os doentes neste importante passo.
Durante a transição de cuidados médicos pediátricos para a idade adulta, questões como a acessibilidade doméstica e tecnologia de assistência, educação e planeamento da formação profissional, e transporte independente para atividades para adultos devem ser tidas em consideração.50
O grupo internacional de peritos em DMD, nas suas orientações de 2018, recomenda o acompanhamento psicossocial do doente e o encaminhamento para um psicólogo ou terapeuta da fala quando necessário em caso de:50
- Incapacidade intelectual
- Transtorno do défice de atenção e hiperatividade
- Desordem do espetro autista
Os doentes com DMD correm um risco acrescido de ansiedade e depressão, especialmente nos principais pontos de transição:50
- A saúde mental e a qualidade de vida devem ser avaliadas em consultas com o especialista neuromuscular e, no mínimo, cada unidade deve ter um plano de prevenção do suicídio. A utilização de intervenções psicofarmacológicas deve ser avaliada para o tratamento de sintomas moderados a graves ou para sintomas mais leves quando os tratamentos farmacológicos não são eficazes.50,51
TRABALHAR EM CONJUNTO PARA OS DOENTES E AS SUAS FAMÍLIAS E PRESTADORES DE CUIDADOS
É possível melhorar a qualidade e a esperança de vida das pessoas que vivem com Duchenne.7,52
Com os cuidados e o acesso a estratégias de gestão adequadas, as equipas de saúde têm sido capazes de o fazer:7,53
- Atrasar a perda de mobilidade
- Reduzir as complicações da doença
- Melhorar a sobrevivência
Para o seu doente e para as pessoas com distrofia muscular, isto pode significar levar uma vida plena e independente até à idade adulta.
O PAPEL DA EQUIPA DE SAÚDE NA GESTÃO DA DISTROFIA MUSCULAR DE DUCHENNE (DMD)
Os cuidados do doente Duchenne podem ser complexos.7
Requer intervenções neuromusculares, respiratórias, cardíacas, ortopédicas, endócrinas, gastrointestinais, psicossociais e reabilitativas, todas elas evoluindo ao longo do curso da doença.7
Como tal, as orientações internacionais para a DMD recomendam uma abordagem coordenada e multidisciplinar dos cuidados de saúde.7,54
Ao ter um grupo de profissionais de saúde numa equipa multidisciplinar, o doente beneficia:7
- Competências e conhecimentos integrados de múltiplas disciplinas
- Um plano de tratamento coordenado e abrangente
A equipa multidisciplinar da DMD:

Adaptado de Birnkrant et al. 2018 (Part1) and Muscular Dystrophy UK
A equipa multidisciplinar é liderada pelo especialista neuromuscular.7,54
PORQUE É IMPORTANTE DIAGNOSTICAR E TRATAR AGORA A DISTROFIA MUSCULAR DE DUCHENNE
Estudos demonstraram que um diagnóstico e cuidados precoces podem ajudar a atrasar a progressão da DMD e minimizar os riscos e complicações da doença.3,6,7,52,55,56
Isto significa que os doentes podem:3,6,7,53,55,56
- Permanecer com mobilidade durante mais tempo
- Preservar a sua função pulmonar e cardíaca
- Viver mais tempo
- Ter uma melhor qualidade de vida
EXISTEM DOIS TIPOS PRINCIPAIS DE TERAPÊUTICA FARMACOLÓGICA PARA A DMD
Agentes que ajudam a controlar os sinais e sintomas da DMD:




* Por favor note que esta não é uma lista exaustiva.
Agentes que tratam a causa subjacente da DMD:
As estratégias terapêuticas incluem:*7,52

Alguns destes tratamentos foram aprovados por entidades reguladoras, enquanto outros ainda se encontram nas fases iniciais de ensaios clínicos ou sob revisão regulatória e poderão estar disponíveis num futuro próximo.7,52
AS ORIENTAÇÕES INTERNACIONAIS PARA OS DOENTES DUCHENNE RECOMENDAM OS GLUCOCORTICOIDES COMO UM DOS PILARES DO TRATAMENTO PARA A DMD
A terapia com glucocorticóides continua a ser um pilar fundamental do tratamento dos doentes de Duchenne e é recomendado que as pessoas continuem a tomar glucocorticóides após a perda de mobilidade.7 Estudos recentes mostraram que quando os glucocorticoides são iniciados em doentes mais jovens, se obtêm maiores benefícios antes de uma deterioração física significativa.7
O tratamento a longo prazo com terapia glucocorticoide demonstrou proporcionar benefícios em comparação com os doentes que não receberam qualquer tratamento glucocorticoide:7



As Orientações Internacionais de Cuidados para a DMD de 2018 recomendam que, após uma consulta inicial com a família, seja realizada uma avaliação dos efeitos secundários e uma consulta de nutrição antes de se iniciar qualquer tratamento com corticosteróides7. Os efeitos secundários do uso prolongado de glicocorticóides podem incluir; aumento de peso e obesidade, acne e verrugas, características cushingoides, atraso do crescimento e puberdade, cataratas, supressão imunitária/adrenal, intolerância à glicose, hipertensão, alterações comportamentais, refluxo gastroesofágico, úlcera péptica, gastrite, osteoporose e mioglobinúria.3,9
Nesta secção encontrará uma extensa coleção de materiais e vídeos sobre a distrofia muscular de Duchenne (DMD) com informações sobre o seu espetro clínico e diagnóstico, bem como sobre os aspetos genéticos desta doença.




50 questões essenciais sobre a Distrofia Muscular de Duchenne.
106 Capítulos com 50 perguntas e respostas.
50 questões essenciais sobre a Distrofia Muscular de Duchenne.
106 Capítulos com 50 perguntas e respostas.

- Consejo General de Colegios Farmacéuticos. Patologías neuromusculares. Avances farmacoterapéuticos recientes. Punto farmacológico. 2020;147:4-10.
- National Institutes of Health. What are the types of muscular dystrophy? Available at: https://www.nichd.nih.gov/health/topics/musculardys/conditioninfo/types.
- Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010 Jan;9(1):77-93. doi: 10.1016/S1474-4422(09)70271-6.
- Theadom A, Rodrigues M, Roxburgh R, Balalla S, Higgins C, Bhattacharjee R, et al. Prevalence of muscular dystrophies: a systematic literature review. Neuroepidemiology. 2014;43(3-4):259-68. doi: 10.1159/000369343.
- Lurio JG, Peay HL, Mathews KD. Recognition and management of motor delay and muscle weakness in children. Am Fam Physician. 2015;91(1):38-44.
- van Ruiten HJ, Straub V, Bushby K, Guglieri M. Improving recognition of Duchenne muscular dystrophy: a retrospective case note review. Arch Dis Child. 2014;99(12):1074-7. doi: 10.1136/archdischild-2014-306366.
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Brumbaugh D, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251-67. doi: 10.1016/S1474-4422(18)30024-3. Erratum in: Lancet Neurol. 2018 Apr 4: PMID: 29395989; PMCID: PMC5869704.
- Brandsema JF, Darras BT. Dystrophinopathies. Semin Neurol. 2015;35(4):369-84. doi: 10.1055/s-0035-1558982.
- Matthews E, Brassington R, Kuntzer T, Jichi F, Manzur AY. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2016;(5):CD003725. doi: 10.1002/14651858.CD003725.pub4.
- Pichavant C, Aartsma-Rus A, Clemens PR, Davies KE, Dickson G, Takeda S, et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol Ther. 2011;19(5):830-40. doi: 10.1038/mt.2011.59.
- Gao QQ, McNally EM. The Dystrophin Complex: Structure, Function, and Implications for Therapy. Compr Physiol. 2015;5(3):1223-39. doi: 10.1002/cphy.c140048.
- Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta. 2007;1772(2):108-17. doi: 10.1016/j.bbadis.2006.05.010.
- Meyers TA, Townsend D. Cardiac Pathophysiology and the Future of Cardiac Therapies in Duchenne Muscular Dystrophy. Int J Mol Sci. 2019;20(17):4098. doi: 10.3390/ijms20174098.
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447(7140):87-91. doi: 10.1038/nature05756.
- Kamdar F, Garry DJ. Dystrophin-Deficient Cardiomyopathy. J Am Coll Cardiol. 2016;67(21):2533-46. doi: 10.1016/j.jacc.2016.02.081.
- Shirokova N, Niggli E. Cardiac phenotype of Duchenne Muscular Dystrophy: insights from cellular studies. J Mol Cell Cardiol. 2013;58:217-24. doi: 10.1016/j.yjmcc.2012.12.009.
- Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT-NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395-402. doi: 10.1002/humu.22758.
- DMD muscular dystrophy (DMD): causes/inheritance. Available at: https://www.mda.org/disease/DMD-muscular-dystrophy/causes-inheritance.
- Goemans N, Buyse G. Current treatment and management of dystrophinopathies. Curr Treat Options Neurol. 2014;16(5):287. doi: 10.1007/s11940-014-0287-4.
- Ferlini A, Neri M, Gualandi F. The medical genetics of dystrophinopathies: molecular genetic diagnosis and its impact on clinical practice. Neuromuscul Disord. 2013;23(1):4-14. doi: 10.1016/j.nmd.2012.09.002.
- Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53(3):145-51. doi: 10.1136/jmedgenet-2015-103387.
- Grounds MD, Terrill JR, Al-Mshhdani BA, Duong MN, Radley-Crabb HG, Arthur PG. Biomarkers for Duchenne muscular dystrophy: myonecrosis, inflammation and oxidative stress. Dis Model Mech. 2020;13(2):dmm043638. doi: 10.1242/dmm.043638.
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev. 2002;82(2):291-329. doi: 10.1152/physrev.00028.2001.
- Humbertclaude V, Hamroun D, Bezzou K, Bérard C, Boespflug-Tanguy O, Bommelaer C, et al. Motor and respiratory heterogeneity in Duchenne patients: implication for clinical trials. Eur J Paediatr Neurol. 2012;16(2):149-60. doi: 10.1016/j.ejpn.2011.07.001.
- Sweeney HL. Developing skeletal muscle MRI/MRS as a biomarker for DMD therapeutic development. Poster presented at the 2014 Annual Connect Conference; 26-28 June, 2014; Chicago, IL.
- Mendell JR, Lloyd-Puryear M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve. 2013;48(1):21-6. doi: 10.1002/mus.23810.
- Griffiths RD, Edwards RH. A new chart for weight control in Duchenne muscular dystrophy. Arch Dis Child. 1988;63(10):1256-8. doi: 10.1136/adc.63.10.1256.
- Ciafaloni E, Fox DJ, Pandya S, Westfield CP, Puzhankara S, Romitti PA, et al. Delayed diagnosis in duchenne muscular dystrophy: data from the Muscular Dystrophy Surveillance, Tracking, and Research Network (MD STARnet). J Pediatr. 2009;155(3):380-5. doi: 10.1016/j.jpeds.2009.02.007.
- Henricson EK, Abresch RT, Cnaan A, Hu F, Duong T, Arrieta A, et al; CINRG Investigators. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other commonly used clinical trial outcome measures. Muscle Nerve. 2013;48(1):55-67. doi: 10.1002/mus.23808.
- Aartsma-Rus A, Hegde M, Ben-Omran T, Buccella F, Ferlini A, Gallano P, et al. Evidence-Based Consensus and Systematic Review on Reducing the Time to Diagnosis of Duchenne Muscular Dystrophy. J Pediatr. 2019;204:305-13.e14. doi: 10.1016/j.jpeds.2018.10.043.
- Romitti PA, Zhu Y, Puzhankara S, James KA, Nabukera SK, Zamba GK, et al; MD STARnet. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics. 2015;135(3):513-21. doi: 10.1542/peds.2014-2044. Erratum in: Pediatrics. 2015;135(5):945.
- Centers for Disease Control and Prevention website. Population-based prevalence of Duchenne and Becker muscular dystrophies in the United States. Available at: https://www.cdc.gov/ncbddd/musculardystrophy/features/key-findings-population-duchenne.html.
- Dent KM, Dunn DM, von Niederhausern AC, Aoyagi AT, Kerr L, Bromberg MB, et al. Improved molecular diagnosis of dystrophinopathies in an unselected clinical cohort. Am J Med Genet A. 2005;134(3):295-8. doi: 10.1002/ajmg.a.30617.
- Vry J, Gramsch K, Rodger S, Thompson R, Steffensen BF, Rahbek J, et al. European Cross-Sectional Survey of Current Care Practices for Duchenne Muscular Dystrophy Reveals Regional and Age-Dependent Differences. J Neuromuscul Dis. 2016;3(4):517-27. doi: 10.3233/JND-160185.
- Wong SH, McClaren BJ, Archibald AD, Weeks A, Langmaid T, Ryan MM, et al. A mixed methods study of age at diagnosis and diagnostic odyssey for Duchenne muscular dystrophy. Eur J Hum Genet. 2015;23(10):1294-300. doi: 10.1038/ejhg.2014.301.
- Darras BT, Urion DK, Ghosh PS. Dystrophinopathies. In: Adam MP, Ardinger HH, Pagon RA, et al, eds. Source Gene Reviews®. Seattle (WA): University of Washington, Seattle; 1993-2019. Published September 5, 2000. Updated April 26, 2018.
- Nascimento Osorio A, Medina Cantillo J, Camacho Salas A, Madruga Garrido M, Vilchez Padilla JJ. Consensus on the diagnosis, treatment and follow-up of patients with Duchenne muscular dystrophy. Neurologia (Engl Ed). 2019;34(7):469-81. English, Spanish. doi: 10.1016/j.nrl.2018.01.001.
- Annexstad EJ, Lund-Petersen I, Rasmussen M. Duchenne muscular dystrophy. Tidsskr Nor Laegeforen. 2014;134:1361-4.
- Nortitz GH, Murphy NA, NEUROMOTOR SCREENING EXPERT PANEL. Motor Delays: Early Identification and Evaluation. Pediatrics. 2013;131(6). Available at: www.pediatrics.org/cgi/content/full/131/6/e2016. Pediatrics. 2017;140(3):e20172081. doi: 10.1542/peds.2017-2081. Erratum for: Pediatrics. 2013;131(6):e2016-27.
- Ardicli, D, Haliloglu, G, Alikasifoglu, M, Topaloğlu H. Diagnostic Pathway to Nonsense Mutation Dystrophinopathy: A Tertiary-Center, Retrospective Experience. Neuropediatrics. 2019;50(1):41-5. doi: 10.1055/s-0038-1675626.
- Counterman KJ, Furlong P, Wang RT, Martin AS. Delays in diagnosis of Duchenne muscular dystrophy: An evaluation of genotypic and sociodemographic factors. Muscle Nerve. 2020;61(1):36-43. doi: 10.1002/mus.26720.
- Wright MA, Yang ML, Parsons JA, Westfall JM, Yee AS. Consider muscle disease in children with elevated transaminase. J Am Board Fam Med. 2012;25(4):536-40. doi: 10.3122/jabfm.2012.04.110183.
- Birnkrant DJ. The American College of Chest Physicians consensus statement on the respiratory and related management of patients with Duchenne muscular dystrophy undergoing anesthesia or sedation. Pediatrics 2009;123(suppl 4):S242-4.
- National Task Force for Early Identification of Childhood Neuromuscular Disorders. Guide for primary care providers. Available at: https://childmuscleweakness.org/wp-content/uploads/2019/05/PrimaryCareProviderPacket.pdf.
- McDonald CM. Clinical approach to the diagnostic evaluation of hereditary and acquired neuromuscular diseases. Phys Med Rehabil Clin N Am. 2012;23(3):495-563. doi: 10.1016/j.pmr.2012.06.011.
- Chakrabarty T, Tirupathi S, Thompson A. How to use: creatine kinase. Arch Dis Child Educ Pract Ed. 2020;105(3):157-63. doi: 10.1136/archdischild-2019-317184.
- National Task Force for Early Identification of Childhood Neuromuscular Disorders. Child Muscle Weakness. 2019. Available at: https://www.childmuscleweakness.org.
- Bianco B, Christofolini DM, Conceição GS, Barbosa CP. Preimplantation genetic diagnosis associated to Duchenne muscular dystrophy. Einstein (Sao Paulo). 2017;15(4):489-91. doi: 10.1590/S1679-45082017RC3994.
- van Dommelen P, van Dijk O, de Wilde JA, Verkerk PH. Early developmental milestones in Duchenne muscular dystrophy. Dev Med Child Neurol. 2020;62(10):1198-204. doi: 10.1111/dmcn.14623.
- Birnkrant DJ, Bushby K, Bann CM, Apkon SD, Blackwell A, Colvin MK, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 3: primary care, emergency management, psychosocial care, and transitions of care across the lifespan. Lancet Neurol. 2018;17(5):445-55. doi: 10.1016/S1474-4422(18)30026-7.
- Birnkrant DJ, Bushby K, Bann CM, Alman BA, Apkon SD, Blackwell A, et al; DMD Care Considerations Working Group. Diagnosis and management of Duchenne muscular dystrophy, part 2: respiratory, cardiac, bone health, and orthopaedic management. Lancet Neurol. 2018;17(4):347-61. doi: 10.1016/S1474-4422(18)30025-5.
- Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat. 2016 Jul 22;12:1795-807. doi: 10.2147/NDT.S93873.
- Gloss D, Moxley RT 3rd, Ashwal S, Oskoui M. Practice guideline update summary: Corticosteroid treatment of Duchenne muscular dystrophy: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology. 2016;86(5):465-72. doi: 10.1212/WNL.0000000000002337.
- Muscular Dystrophy UK. Care Advisors and Clinical Nurse Specialists. http://www.musculardystrophyuk.org/get-the-right-care-and-support/people-and-places-to-help-you/professionals-and-organisations/care-advisors/
- Merlini L. A 19-year-old ambulant Duchenne patient with stunted growth on long-term corticosteroids. Neuromuscul Disord. 2014;24(5):417-8. doi: 10.1016/j.nmd.2014.02.006.
- Merlini L, Gennari M, Malaspina E, Cecconi I, Armaroli A, Gnudi S, et al. Early corticosteroid treatment in 4 Duchenne muscular dystrophy patients: 14-year follow-up. Muscle Nerve. 2012;45(6):796-802. doi: 10.1002/mus.23272.
- McDonald CM, Henricson EK, Abresch RT, Duong T, Joyce NC, Hu F, et al; CINRG Investigators. Long-term effects of glucocorticoids on function, quality of life, and survival in patients with Duchenne muscular dystrophy: a prospective cohort study. Lancet. 2018;391(10119):451-61. doi: 10.1016/S0140-6736(17)32160-8.