Deficiência da DAA
A deficiência da descarboxilase dos L-aminoácidos aromáticos (d-DAA) é uma doença hereditária que afeta a síntese de neurotransmissores e causa uma ampla variedade de sintomas debilitantes, como disfunção motora e autonómica e atraso no desenvolvimento, afetando as atividades diárias.
A deficiência da DAA é causada por variantes patogénicas no gene dopa descarboxilase DDC, que codifica a enzima DAA.2,3
São atualmente conhecidas cerca de 100 variantes patogénicas homozigóticas ou heterozigóticas compostas em doentes com deficiência da DAA. 3,4,7,8
A variante mais frequente é IVS6+4A>T; contudo, esta mutação está presente em menos de metade dos casos confirmados de deficiência da DAA.2,3
Não existe uma correlação direta entre a gravidade da deficiência da DAA e as variantes específicas conhecidas; a maioria das variantes resulta num quadro clínico inicial variável.
Número e localização das variantes patogénicas no gene DDC3
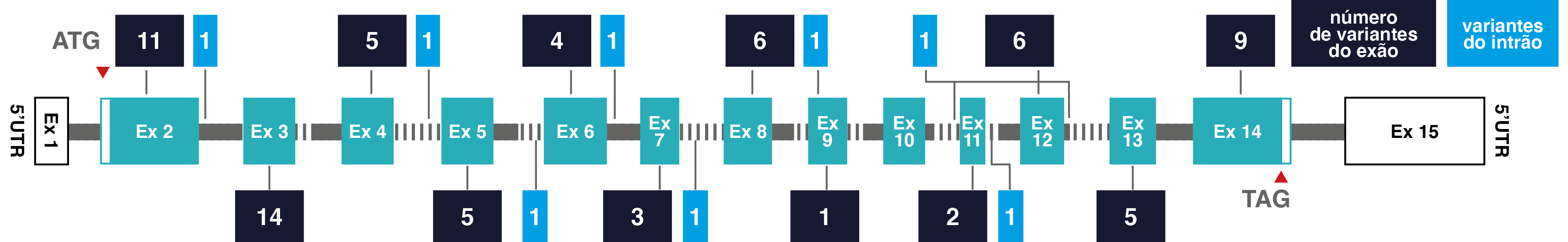
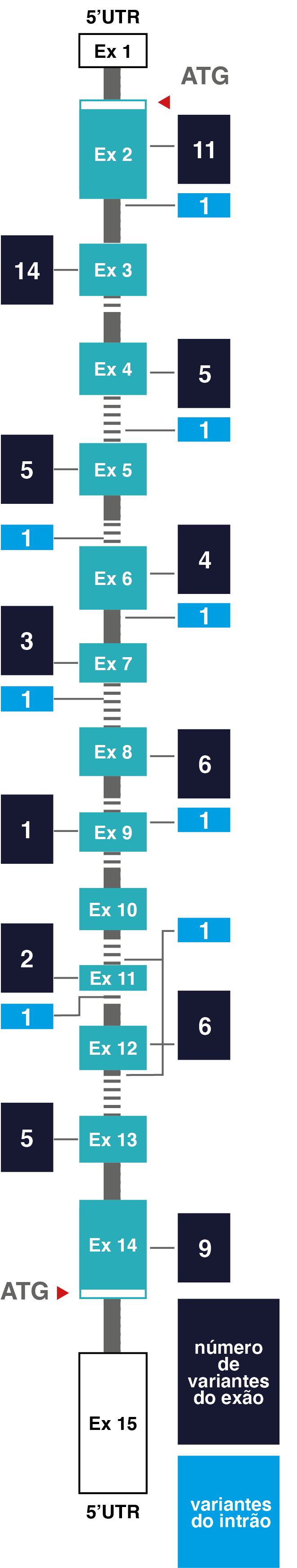
Ex, exão; 3’UTR, região não traduzida em 3’; 5’UTR, região não traduzida em 5’.
A ausência de atividade enzimática DAA inibe a síntese dos neurotransmissores monoamina e causa os principais sinais e sintomas da deficiência da DAA.3
A ausência de atividade enzimática DAA resulta na acumulação de serotonina e precursores de dopamina (5-HTP, L-DOPA, 3-OMD), bem como na diminuição dos níveis dos metabolitos de serotonina e da catecolamina (5-HIAA, HVA e MHPG).2
Isto resulta numa grave deficiência em dopamina, serotonina e outras catecolaminas, tais como noradrenalina e adrenalina.2,3
A ausência destes neurotransmissores inibe a sinalização neuronal no sistema nervoso central, o que influencia o desenvolvimento motor, o comportamento e a função autonómica.3
Os baixos níveis de neurotransmissores contribuem para o desenvolvimento dos sintomas sentidos pelos doentes com deficiência da DAA.6
A deficiência da DAA apresenta uma grande variedade de manifestações clínicas.3,6
Os doentes com deficiência da DAA apresentam um largo espetro de fenótipos com disfunção motora, atraso no desenvolvimento e dificuldade em alcançar marcos de desenvolvimento fundamentais.2-9 Os doentes com deficiência da DAA podem também apresentar disautonomia.2,3
Sintomas tais como hipotonia, crises oculogíricas, atraso no desenvolvimento motor e sinais disautonómicos estão consistentemente associados à deficiência da DAA.3 No entanto, estes sintomas podem variar de leves a graves, o que explica a grande variabilidade na apresentação clínica.3 Os doentes com as formas mais graves de deficiência da DAA podem nunca alcançar as metas de desenvolvimento.9
Revisão bibliográfica de 117 doentes com deficiência da DAA,
descrevendo as características dos doentes, quando disponíveis.2
Apresentação clínica em doentes com deficiência da DAA
Os sintomas de deficiência da DAA podem aparecer numa idade precoce; de acordo com as orientações de consenso publicadas.2,3
-
Os sintomas aparecem geralmente no primeiro ano de vida e a idade média
dos doentes que apresentam sintomas é de 2,7 meses (n=68).
-
Apesar desta idade precoce de início dos sintomas, a idade média no diagnóstico
é de 3,5 anos *(n=87).
A deficiência da DAA pode apresentar sinais e sintomas semelhantes aos de outras doenças neurológicas (por exemplo, paralisia cerebral, epilepsia) e de algumas perturbações comportamentais.3
Os sinais e sintomas de deficiência da DAA podem sobrepor-se aos de outras doenças.3
Apresentação clínica da deficiência da DAA comparada com doenças com uma apresentação semelhante2,3,10
Sinais e sintomas da deficiência da DAA2,3,10
- Hipotonia
- Hipertonia
- Discinesia
- Distonia
- Hipercinesia
Doenças com sinais e sintomas semelhantes
Transtornos do movimento ou Paralisia cerebral 2,11,12
Sinais e sintomas da deficiência da DAA2,3,10
- Convulsões
- Movimentos oculares anómalos (crises oculogíricas)
- Distonia paroxística
Doenças com sinais e sintomas semelhantes
Epilepsia12,14
(crises oculogíricas observadas como convulsões)
Sinais e sintomas da deficiência da DAA2,3,10
- Irritabilidade
- Disforia
- Choro excessivo
- Problemas da fala
- Incapacidade intelectual
Doenças com sinais e sintomas semelhantes
Distúrbios de
comportamento/autismo15,16
A deficiência da DAA foi documentada em todo o mundo1 e afeta doentes de diferentes origens, tais como:2,3
Asiáticos
Caucasianos
Árabes
Iranianos
Judeus
O termo prevalência de nascimento ou prevalência de recém-nascidos é preferido ao termo incidência, que é o número de novos casos da doença num determinado momento. Prevalência é o número de pessoas vivas com uma doença num determinado momento e é um termo mais preciso utilizado para as doenças genéticas em que o genótipo está presente à nascença.17
Como é o caso de muitas outras doenças raras que são subdiagnosticadas, é difícil estimar a prevalência da deficiência da DAA. As estimativas de prevalência de nascimentos variam entre 1:32.000 e 1:182.000, depedendo da área geográfica.3,18,19
Análise bibliográfica sobre a prevalência da deficiência da DAA ao nascimento3,18-20
a A prevalência ao nascimento tem por base os índices de prevalência numa população de risco (doentes que apresentam deficiências neurológicas de origem desconhecida). b Prevalência ao nascimento estimada utilizando as frequências das variantes patogénicas dos alelos do gene DDC.
Os doentes que apresentem os principais sinais e sintomas de deficiência da DAA devem ser testados quanto à presença de uma doença de neurotransmissores.
Diagnóstico da deficiência da DAA em doentes com suspeita de uma doença da neurotransmissão2
+
Atraso no desenvolvimento motor
+
RM estruturalmente normal*
- Crises oculogíricas
- Distonia
- Hipocinesia e/ou tradicinesia
V
Temperatura instável
Congestão nasal
Sudorese excessiva
RM, ressonância magnética
* Os doentes com deficiência da DAA podem apresentar alterações heterogéneas na RM sem relação com as manifestações clínicas2.
Figura adaptada de Himmelreich N, et al.3
Os 3 exames de diagnóstico fundamentais para confirmar a deficiência da DAA são2,3:
- As atuais orientações de consenso recomendam testes genéticos para confirmar o diagnóstico de deficiência da DAA2.
Resultados expectáveis em doentes com deficiência da DAA:
- Aumento da concentração de L-DOPA, 3-OMD e 5-HTP.
- Diminuição das concentrações de HVA, 5-HIAA e MHPG.
- Níveis normais de pterina.
observa-se uma atividade reduzida da enzima DAA no plasma.
Os 3 exames de diagnóstico essenciais para confirmar a presença de deficiência da DAA2,3
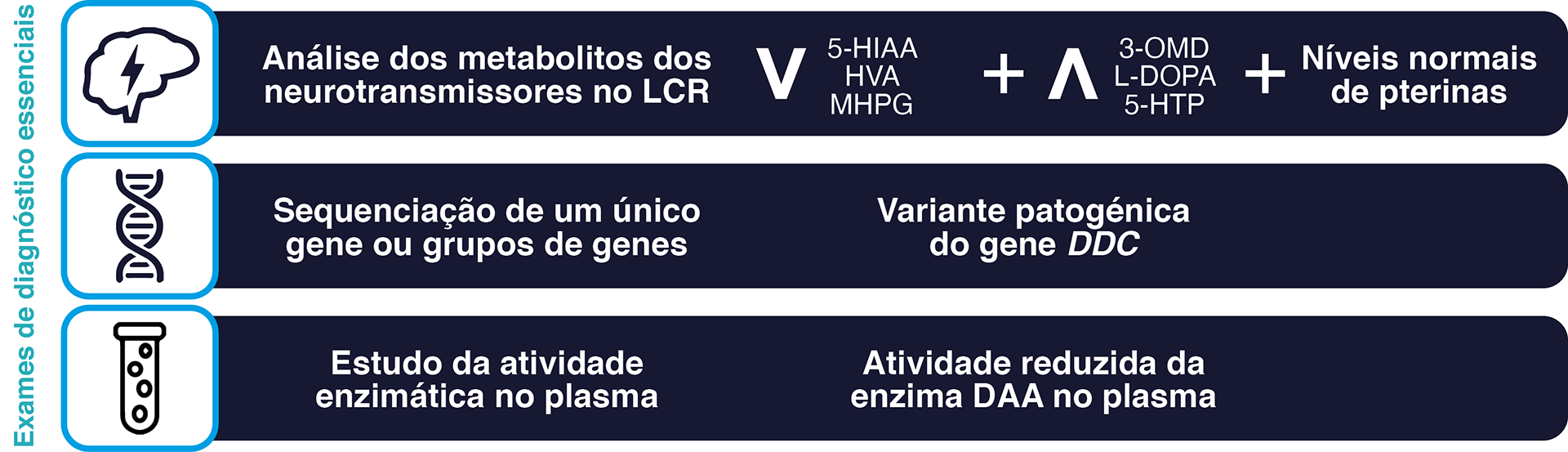
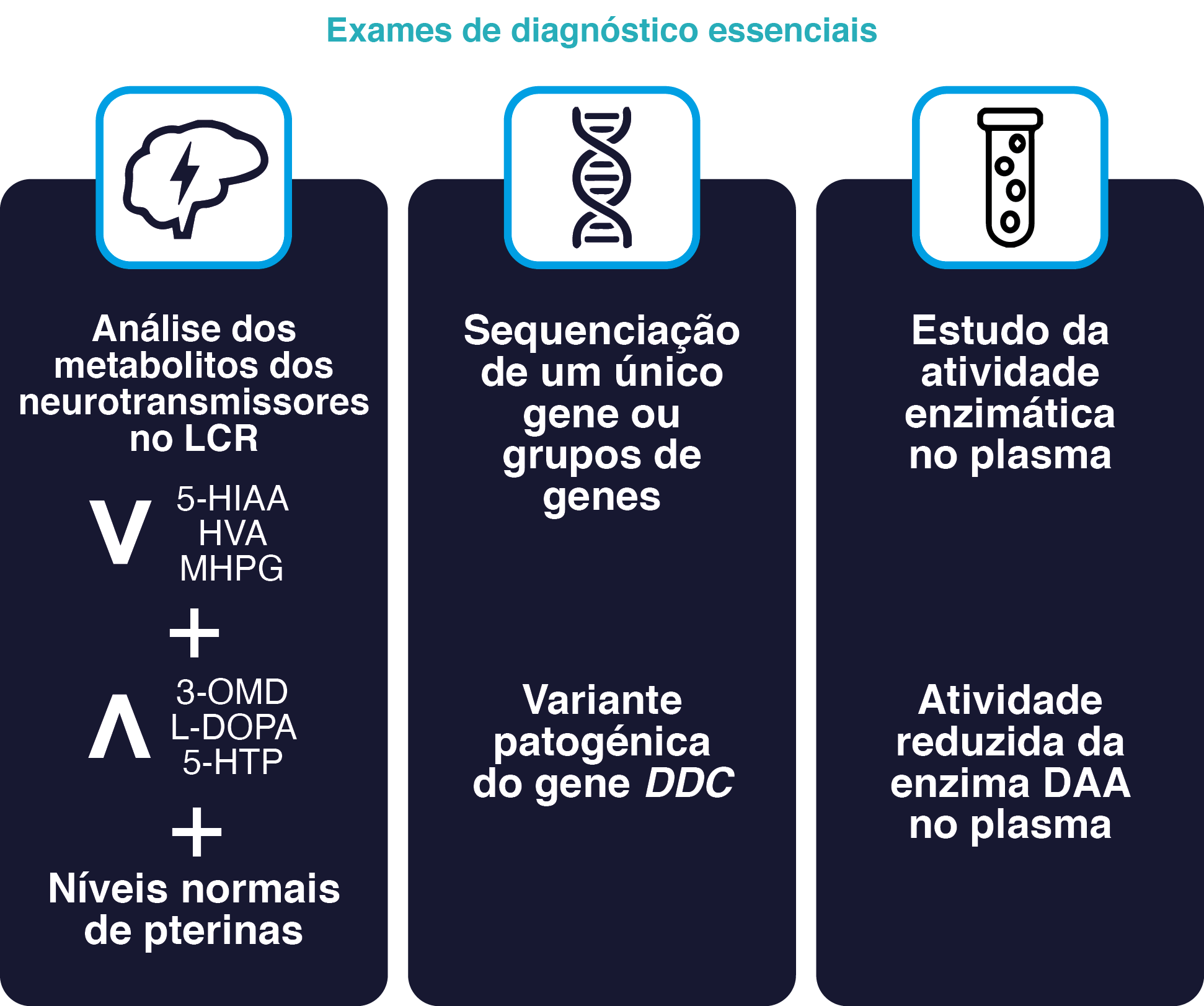
3-OMD, 3-O-metildopa; 5-HIAA, ácido 5-hidroxiindolacético; 5-HTP, 5-hidroxitriptofano; DDC, dopa descarboxilase, HVA, ácido homovanílico; LCR, líquido cefalorraquidiano; L-dopa; levodopa; MHPG, 3-metoxi-4-hidroxifenilglicol.
Figura adaptada de Wassenberg T, et al.2 e de Himmelreich N, et al.3
Estão disponíveis outros exames para diagnóstico da deficiência da DAA, tais como a análise da concentração de 3-OMD no sangue ou plasma e a análise de ácidos orgânicos na urina.2,19-23
Concentração de 3-OMD ou de ácido vanilíco como ferramentas adicionais para o diagnóstico da deficiência da DAA2,3,19-25

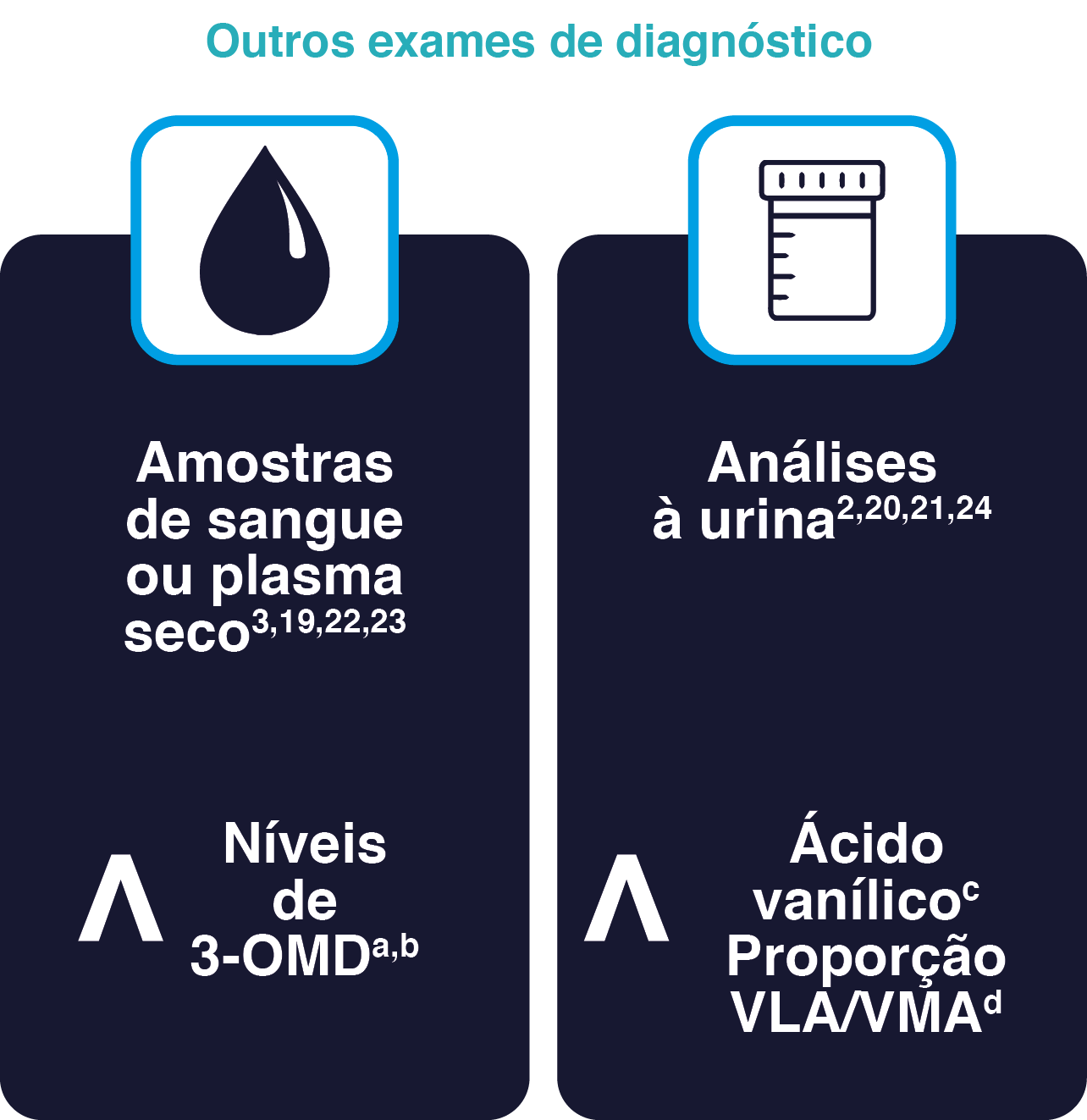
3-OMD, 3-O-metildopa; VLA, ácido vanilático; VMA, ácido vanilmandélico.
a Níveis elevados de 3-OMD são também observados na deficiência de piridoxal 5'-fosfato oxidase, embora estes doentes tenham uma encefalopatia epiléptica grave, que tem uma apresentação diferente da deficiência da DAA.24
b Os doentes expostos a medicamentos relacionados com L-DOPA podem ter níveis elevados de 3-OMD, tanto os doentes que tomam medicamentos como os recém-nascidos de mães que tomam medicamentos que afetam o sistema dopaminérgico. Isto deve ser tido em conta ao interpretar os resultados de rastreio dos recém-nascidos. Além disso, deve ser evitado o uso destes medicamentos antes dos testes de urina.221,22
c O aumento do ácido vanilático é frequentemente subtil e pode passar despercebido se não for especificamente pesquisado num laboratório especializado.2
d Não faz parte das orientações de consenso, mas pode ser útil para o rastreio.21 A medida do rácio VLA/VMA em 10095 amostras de controlo foi de 0,007 (DP=0,37, intervalo 0,0-23,24), enquanto que a medida do rácio em 14 análises de nove doentes individuais com deficiência da DAA foi de 23,16 (DP=22,83, intervalo 0,97-74,1), representando um aumento de aproximadamente 350 vezes. Portanto, a relação VLA/VMA permite a identificação fiável de doentes com deficiência da DAA, especialmente na faixa etária mais jovem, uma vez que diminui com a idade.25
A análise de amostras de sangue seco para concentração 3-OMD é um teste simples, rápido e minimamente invasivo que pode ser utilizado para confirmar a presença de deficiência da DAA.5,19,22
Pode ser incluído no rastreio neonatal19,22 para promover o diagnóstico precoce da deficiência da DAA e assim reduzir o atraso no diagnóstico da patologia5,19.
Se a concentração de 3-OMD ou de ácido vanílico for elevada, devem ser efetuados exames de diagnóstico essenciais (estudo da atividade plasmática, análise do LCR e testes genéticos) para confirmar o diagnóstico.2
Os sintomas da deficiência da descarboxilase dos L-aminoácidos aromáticos (d-DAA) sobrepõem-se marcadamente aos sintomas observados noutras doenças neurológicas, o que pode levar a um diagnóstico erróneo. Os infográficos seguintes mostram as manifestações clínicas da deficiência da DAA e outras doenças comuns, tais como paralisia cerebral, epilepsia, doenças neuromusculares e mitocondriopatias, bem como as principais características que nos permitirão obter um diagnóstico preciso e atempado.2,6,26-28
Paralisia cerebral
A paralisia cerebral (PC) descreve um grupo de perturbações permanentes do desenvolvimento neuromotor que causam limitações de atividade. São atribuídos a perturbações não progressivas no desenvolvimento cerebral do feto ou da criança, acompanhadas de um atraso no desenvolvimento29,30. É a deficiência física mais comum em crianças, com uma prevalência entre 1,7 e 3,1 casos por 1.000 habitantes nos países desenvolvidos.31
Existem diferentes tipos de PC, que são classificados de acordo com a área do sistema nervoso central afetada e as manifestações clínicas:32,33
Muitas das manifestações da PC durante a infância são semelhantes às observadas em doentes com perturbações do neurotransmissor monoamina, tais como deficiência da DAA, que podem muitas vezes conduzir a diagnósticos e tratamentos erróneos.27
Os principais sintomas da deficiência da DAA que normalmente não são vistos na PC são convulsões oculogíricas e flutuação diurna dos sintomas motores. Nestes casos, são necessários exames apropriados, tais como a análise dos neurotransmissores no líquido cefalorraquidiano, para se obter um diagnóstico clínico preciso.30,34
Além disso, os doentes com doenças que mimetizam a PC apresentam outros sintomas que diferem da PC clássica e podem fornecer uma pista para o diagnóstico, tais como vários sinais disautonómicos, por exemplo, suor excessivo (diaforese), temperatura instável, descarga nasal e orofaríngea abundante, distúrbios do movimento intestinal (dismotilidade gastrointestinal) e distúrbios do sono.30
Em doentes pediátricos ou adultos com uma perturbação motora não diagnosticada compatível com a PC, o teste de primeira linha é uma RM ao cérebro. Estudos de neuroimagem mostram claramente se a principal causa dos sintomas neurológicos é uma malformação cerebral. Para além das malformações cerebrais, estes estudos podem mostrar resultados normais ou lesões específicas características de uma doença genética ou grupo de doenças, indicando a necessidade de mais exames complementares.30
Sintomas comuns e distintivos da PC e da deficiência da DAA*

† As convulsões são relativamente raras na PC e raras na deficiência da DAA, embora tenham sido observadas em ambos os casos. As convulsões na PC correspondem principalmente a um subgrupo generalizado ou parcial.12,13
‡ A flutuação diurna nem sempre é um sinal de deficiência da DAA, embora seja observada de forma consistente.11
Esta não é uma lista completa
voltar
Epilepsia
A epilepsia é uma doença neurológica crónica caracterizada por convulsões recorrentes e imprevisíveis resultantes de uma atividade neuronal anormal excessiva ou síncrona no cérebro.38,39
É uma das doenças neurológicas mais comuns em todo o Mundo, afetando cerca de 50 milhões de pessoas de todas as idades.39
As crises epilépticas dividem-se em três categorias:40
Episódios paroxísticos frequentemente vividos por doentes com deficiência da DAA, tais como convulsões oculogíricas, postura tónica ou distónica dos membros, mioclonus e coreia, podem ser confundidos com convulsões e, portanto, erroneamente diagnosticados como tal.2,6,41-43 Por esta razão, antes da confirmação do diagnóstico de deficiência da DAA, os doentes recebem múltiplos tratamentos com medicamentos antiepiléticos sem que seja observada qualquer resposta.43 Em particular, os resultados do EEG de doentes com deficiência da DAA e doentes com epilepsia diferem frequentemente, pelo que é essencial efetuar uma análise do EEG para distinguir entre doentes com episódios paroxísticos e doentes com epilepsia.41
As crises epiléticas são raras em doentes com deficiência da DAA5, embora tenham sido notificados alguns casos10,41,44-46. Com base num número limitado de casos clínicos publicados, as convulsões tendem a ocorrer nos primeiros anos de vida, não são severas, são geralmente TCG ou convulsões focais complexas e, uma vez documentadas, são bem controladas com a administração de medicamentos antiepiléticos convencionais.10,41,44-46
A diferenciação das crises epiléticas de episódios paroxísticos não epiléticos pela análise EEG é essencial para evitar um mau diagnóstico da deficiência da DAA como epilepsia e para assegurar que os doentes recebem o tratamento adequado.41
Sinais diferenciadores de crises epiléticas e de deficiência da DAA

* As crises oculogíricas foram observadas em 97% (n=30/31) dos doentes com deficiência da DAA entre os 2 e os 12 anos de idade47
† Excecionalmente, a distonia pode ser um sinal em doenças epiléticas raras48
‡ Os doentes com deficiência da DAA normalmente não sofrem convulsões tónicas generalizadas, embora estas tenham sido observadas num pequeno número de doentes com deficiência da DAA que também foram diagnosticados com epilepsia42
voltar
Perturbações neuromusculares
As perturbações neuromusculares são um vasto grupo de doenças genéticas heterogéneas que afetam os músculos ou o sistema nervoso periférico. 49-51 Seguem-se alguns tipos de distúrbios neuromusculares49,52
- Doença de Charcot-Marie-Tooth
- Esclerose múltipla
- Distrofia muscular
- Miastenia gravis
- Miopatia
- Miosite (polimiosite e dermatomiosite)
- Neuropatia periférica
- Atrofia muscular espinhal
O problema pode ter origem em corpos celulares (ELA ou ganglionopatias sensoriais), axónios (neuropatias periféricas axonais ou plexopatias braquiais), células de Schwann (poliradiculoneuropatia desmielinizante inflamatória crónica), junções neuromusculares (miastenia gravis ou síndrome miasténica de Lambert-Eaton), tecido muscular (miopatia inflamatória ou distrofia muscular) ou uma combinação das anteriores. As perturbações neuromusculares variam no desenvolvimento e gravidade da doença54 dependendo da região do corpo afetada, mas os sintomas mais comuns são fraqueza muscular, marcha alterada ou diminuída, contraturas articulares, deformações ósseas, perceção sensorial alterada (neuropatias) e insuficiência respiratória49. Os doentes com função muscular normal intermédia ou distúrbios neuromusculares persistentes podem apresentar mialgia, rabdomiólise, fraqueza e fadiga.49
Considerando que 50% dos doentes com deficiência da DAA têm perturbações do movimento (hipocinesia, coreia, distonia, balismo, discinesia, tremor, mioclonismo), a deficiência da DAA pode ser confundida com diferentes perturbações neuromusculares, tais como miastenia gravis congénita, o que pode levar a um diagnóstico erróneo.27,28
Sintomas comuns e distintivos de perturbações neuromusculares e da deficiência da DAA

voltar
Mitocondriopatias
As mitocondriopatias são um grupo heterogéneo de doenças causadas por perturbações da cadeia respiratória mitocondrial.56 A prevalência de mitocondriopatias é estimada em pelo menos 20 por 100.000 habitantes.57
As manifestações mais comuns de mitocondriopatias são a ptose, oftalmoplegia externa, miopatia proximal e intolerância ao exercício, cardiomiopatia, surdez neurossensorial, atrofia óptica, retinopatia pigmentar e diabetes mellitus.56
Principais manifestações clínicas das mitocondriopatias56

CPEO, oftalmoplegia externa progressiva crónica; LCR, líquido cefalorraquidiano; LHON, neuropatia óptica hereditária de Leber; MELAS, encefalomiopatia mitocondrial com acidose lática e episódios semelhantes a um acidente vascular cerebral; MEMSA, epilepsia mioclónica com ataxia sensorial; MERRF, epilepsia mioclónica com fibras vermelhas rasgadas; MIRAS, síndrome da ataxia mitocondrial recessiva; NARP, fraqueza neurogénica com ataxia e retinite pigmentosa; SANDO, neuropatia ataxica-disartríase sensorial; SCAE, ataxia espinocerebelar com epilepsia; KSS, síndrome de Kearns-Sayre; LIMM, miopatia mitocondrial letal do lactente; SNA, síndrome de neuropatia ataxia; SNA, síndrome de neuropatia ataxia; SCAE, síndrome de ataxia espinocerebelar com epilepsia.
* Também conhecido como MIRAS e SCAE.
Nas crianças, manifestações clínicas como a síndrome hipocinética rígida e a distonia generalizada podem ser confundidas com as observadas na deficiência da DAA e outros distúrbios neurotransmissores pediátricos, tornando o diagnóstico baseado apenas na apresentação clínica difícil.58 A presença de crises oculogíricas pode ajudar a distinguir a deficiência da DAA de uma mitocondriopatia, no entanto, não é suficiente para confirmar o diagnóstico, uma vez que as crises oculogíricas também são observadas em algumas mitocondriopatias.59 Portanto, os testes genéticos moleculares são necessários para confirmar ou descartar o diagnóstico de deficiência da DAA.2
Sintomas comuns e distintivos de mitocondriopatias e da deficiência da DAA

*As apreensões são raras na deficiência da DAA, embora tenham sido relatados alguns casos.
†Embora tenham sido observados
casos de crises oculogíricas, estas não são típicas de mitocondriopatias.
voltar
Em doentes com distúrbios de neurotransmissores, tais como a deficiência da DAA, os sintomas podem não ser específicos e o diagnóstico é muitas vezes tardio.61 Como consequência, os doentes e prestadores de cuidados de saúde percorrem um longo caminho até ao diagnóstico e frequentemente recebem diagnósticos errados, por exemplo, paralisia cerebral, miastenia gravis ou perturbações convulsivas, antes de se encontrar o diagnóstico correto.61 Estes erros ou atrasos no diagnóstico podem causar o agravamento do estado do doente.61
Os doentes podem apresentar outras complicações, tais como problemas cardíacos e ortopédicos. 2 Para além do atraso no crescimento, estes doentes estão mais propensos a infeções, geralmente, como resultado de complicações decorrentes de problemas de alimentação e de deglutição, mobilidade reduzida e admissões recorrentes.2
A necessidade contínua de assistência física e os problemas comportamentais dos doentes com deficiência da DAA representam um grande encargo para os cuidadores.2,3
Embora não haja muita informação disponível sobre a qualidade de vida dos cuidadores de doentes com deficiência da DAA, podemos ter uma ideia a partir das experiências dos cuidadores de outras doenças neurológicas pediátricas com sintomas semelhantes.2
- Os resultados de inquéritos a cuidadores de doentes com paralisia cerebral mostram que os cuidadores tendem a ter níveis mais elevados de stress e depressão e uma qualidade de vida mais baixa do que os pais de crianças saudáveis.62 Os fatores mais frequentemente citados como a causa destes efeitos são os problemas cognitivos e comportamentais da criança, a baixa auto-eficácia do prestador de cuidados e a falta de apoio social.62
No caso de doenças de neurotransmissores, tais como deficiência da DAA, os erros frequentes e o diagnóstico tardio são uma causa de stress para os prestadores de cuidados e representam uma carga económica considerável para o sistema de saúde.61
Em muitos doentes com deficiência da DAA, os atrasos no desenvolvimento, deficiências motoras profundas e outras comorbilidades psiquiátricas ou de desenvolvimento neurológico que requerem cuidados ao longo da vida, como noutras doenças crónicas como a epilepsia, colocam um pesado encargo aos prestadores de cuidados e prejudicam a sua qualidade de vida.2,63,64
*Estudo retrospetivo, descritivo e monocêntrico de doentes diagnosticados com deficiência da DAA no Hospital Universitário Nacional de Taiwan entre 2004 e 2016.
Nesta secção encontrará uma vasta compilação de materiais e vídeos sobre a deficiência da descarboxilase dos L-aminoácidos aromáticos (d-DAA), desde informação sobre o seu espetro clínico e diagnóstico diferencial, bem como aspetos bioquímicos e genéticos desta doença.





1. García-Cazorla A, Ormazábal A, Artuch R, Pérez-Dueñas B, López-Casas J, Fernández-Álvarez E, et al. Errores congénitos de los neurotransmisores en Neuropediatría. Rev Neurol 2005;41(02):99-108. doi: 10.33588/rn.4102.2004377.
2. Wassenberg T, Molero-Luis M, Jeltsch K, Hoffmann GF, Assmann B, Blau N, et al. Consensus guideline for the diagnosis and treatment of aromatic l-amino acid decarboxylase (AADC) deficiency. Orphanet J Rare Dis. 2017;12(1):12. doi: 10.1186/s13023-016-0522-z.
3. Himmelreich N, Montioli R, Bertoldi M, Carducci C, Leuzzi V, Gemperle C, et al. Aromatic amino acid decarboxylase deficiency: Molecular and metabolic basis and therapeutic outlook. Mol Genet Metab. 2019;127(1):12-22. doi: 10.1016/j.ymgme.2019.03.009.
4. Dai L, Ding C, Fang F. A novel DDC gene deletion mutation in two Chinese mainland siblings with aromatic l-amino acid decarboxylase deficiency. Brain Dev. 2019;41(2):205-9. doi: 10.1016/j.braindev.2018.08.003.
5. Chen PW, Lee NC, Chien YH, Wu JY, Wang PC, Hwu WL. Diagnosis of aromatic L-amino acid decarboxylase deficiency by measuring 3-O-methyldopa concentrations in dried blood spots. Clin Chim Acta. 2014;431:19-22. doi: 10.1016/j.cca.2014.01.034.
6. Brun L, Ngu LH, Keng WT, Ch'ng GS, Choy YS, Hwu WL, et al. Clinical and biochemical features of aromatic L-amino acid decarboxylase deficiency. Neurology. 2010;75(1):64-71. doi: 10.1212/WNL.0b013e3181e620ae. Erratum in: Neurology. 2010;75(6):576. Dosage error in article text.
7. Monteleone B, Hyland K. Case report: discovery of 2 gene variants for aromatic L-amino acid decarboxylase deficiency in 2 African American siblings. BMC Neurol. 2020;20(1):12. doi: 10.1186/s12883-019-1596-8.
8. Micallef J, Stockler-Ipsiroglu S, van Karnebeek CD, Salvarinova-Zivkovic R, Horvath G. Recurrent Dystonic Crisis and Rhabdomyolysis Treated with Dantrolene in Two Patients with Aromatic L-Amino Acid Decarboxylase Deficiency. Neuropediatrics. 2020;51(3):229-32. doi: 10.1055/s-0039-3402010.
9. Hwu WL, Chien YH, Lee NC, Li MH. Natural History of Aromatic L-Amino Acid Decarboxylase Deficiency in Taiwan. JIMD Rep. 2018;40:1-6. doi: 10.1007/8904_2017_54.
10. Manegold C, Hoffmann GF, Degen I, Ikonomidou H, Knust A, Laass MW, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, drug therapy and follow-up. J Inherit Metab Dis. 2009;32(3):371-80. doi: 10.1007/s10545-009-1076-1.
11. Blackburn JS, Mink JW, Augustine EF. Pediatric movement disorders: Five new things. Neurol Clin Pract. 2012;2(4):311-8. doi: 10.1212/CPJ.0b013e318278bf06.
12. Hallman-Cooper JL, Rocha Cabrero F. Cerebral Palsy. [Updated 2021 Apr 28]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2021 Jan. www.ncbi.nlm.nih.gov/books/NBK538147/
13. Menghi V, Bisulli F, Tinuper P, Nobili L. Sleep-related hypermotor epilepsy: prevalence, impact and management strategies. Nat Sci Sleep. 2018;10:317-26. doi: 10.2147/NSS.S152624.
14. Shibata M, Kato T, Yoshida T, Saito K, Awaya T, Heike T. Paroxysmal gaze deviations as the sole manifestation of occipital lobe epilepsy. Seizure. 2013;22(10):913-5. doi: 10.1016/j.seizure.2013.06.012.
15. DeFilippis M, Wagner KD. Treatment of Autism Spectrum Disorder in Children and Adolescents. Psychopharmacol Bull. 2016;46(2):18-41.
16. Kim JS. Excessive crying: behavioral and emotional regulation disorder in infancy. Korean J Pediatr. 2011;54(6):229-33. doi: 10.3345/kjp.2011.54.6.229.
17. Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, et al. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review. Orphanet J Rare Dis. 2017;12(1):124. doi: 10.1186/s13023-017-0671-8.
18. Hyland K, Reott M. Prevalence of Aromatic l-Amino Acid Decarboxylase Deficiency in At-Risk Populations. Pediatr Neurol. 2020;106:38-42. doi: 10.1016/j.pediatrneurol.2019.11.022.
19. Chien YH, Chen PW, Lee NC, Hsieh WS, Chiu PC, Hwu WL, et al. 3-O-methyldopa levels in newborns: Result of newborn screening for aromatic l-amino-acid decarboxylase deficiency. Mol Genet Metab. 2016;118(4):259-63. doi: 10.1016/j.ymgme.2016.05.011.
20. Hyland K, Clayton PT. Aromatic L-amino acid decarboxylase deficiency: diagnostic methodology. Clin Chem. 1992;38(12):2405-10.
21. Lee HC, Lai CK, Yau KC, Siu TS, Mak CM, Yuen YP, et al. Non-invasive urinary screening for aromatic L-amino acid decarboxylase deficiency in high-prevalence areas: a pilot study. Clin Chim Acta. 2012;413(1-2):126-30. doi: 10.1016/j.cca.2011.09.008.
22. Brennenstuhl H, Kohlmüller D, Gramer G, Garbade SF, Syrbe S, Feyh P, et al. High throughput newborn screening for aromatic ʟ-amino-acid decarboxylase deficiency by analysis of concentrations of 3-O-methyldopa from dried blood spots. J Inherit Metab Dis. 2020;43(3):602-10. doi: 10.1002/jimd.12208.
23. Atwal PS, Donti TR, Cardon AL, Bacino CA, Sun Q, Emrick L, et al. Aromatic L-amino acid decarboxylase deficiency diagnosed by clinical metabolomic profiling of plasma. Mol Genet Metab. 2015;115(2-3):91-4. doi: 10.1016/j.ymgme.2015.04.008.
24. Mills PB, Surtees RA, Champion MP, Beesley CE, Dalton N, Scambler PJ, et al. Neonatal epileptic encephalopathy caused by mutations in the PNPO gene encoding pyridox(am)ine 5'-phosphate oxidase. Hum Mol Genet. 2005;14(8):1077-86. doi: 10.1093/hmg/ddi120.
25. Brennenstuhl H, Garbade SF, Okun JG, Feyh P, Hoffmann GF, Langhans CD, et al. Semi-quantitative detection of a vanillactic acid/vanillylmandelic acid ratio in urine is a reliable diagnostic marker for aromatic L-amino acid decarboxylase deficiency. Mol Genet Metab. 2020;131(1-2):163-70. doi: 10.1016/j.ymgme.2020.07.001.
26. Krigger KW. Cerebral palsy: an overview.Am FamPhysician. 2006;73(1):91-100.
27. Ng J, Papandreou A, Heales SJ, Kurian MA. Monoamine neurotransmitter disorders — clinical advances and future Perspectives. Nat Rev Neurol. 2015;11(10):567-84.
28. Kurian MA, Dale RC. Movement disorders presenting in childhood. Continuum (Minneap Minn). 2016;22(4 Movement Disorders):1159-85.
29. Novak I, Morgan C, Adde L, Blackman J, Boyd RN, Brunstrom-Hernandez J, et al. Early, Accurate Diagnosis and Early Intervention in Cerebral Palsy: Advances in Diagnosis and Treatment. JAMA Pediatr. 2017;171(9):897-907. doi: 10.1001/jamapediatrics.2017.1689. Erratum in: JAMA Pediatr. 2017;171(9):919.
30. Pearson TS, Pons R, Ghaoui R, Sue CM. Genetic mimics of cerebral palsy. Mov Disord. 2019;34(5):625-36. doi: 10.1002/mds.27655.
31. Monbaliu E, Himmelmann K, Lin JP, Ortibus E, Bonouvrié L, Feys H, et al. Clinical presentation and management of dyskinetic cerebral palsy. Lancet Neurol. 2017;16(9):741-9. doi: 10.1016/S1474-4422(17)30252-1.
32. Cerebral Palsy Guidance. Cerebral Palsy Symptoms. Available from: https://www.cerebralpalsyguidance.com/cerebral-palsy/symptoms/.
33. Cerebral Palsy (CP) Syndromes. (n.d.). Merck Manuals Professional Edition. Available from: https://www.merckmanuals.com/professional/pediatrics/neurologic-disorders-in-children/cerebral-palsy-cp-syndromes. 34. Kurian MA, Gissen P, Smith M, Heales S Jr, Clayton PT. The monoamine neurotransmitter disorders: an expanding range of neurological syndromes. Lancet Neurol. 2011;10(8):721-33. doi: 10.1016/S1474-4422(11)70141-7.
35. Zouvelou V, Yubero D, Apostolakopoulou L, Kokkinou E, Bilanakis M, Dalivigka Z, et al. The genetic etiology in cerebral palsy mimics: The results from a Greek tertiary care center. Eur J Paediatr Neurol. 2019;23(3):427-37. doi: 10.1016/j.ejpn.2019.02.001.
36. Gururaj AK, Sztriha L, Bener A, Dawodu A, Eapen V. Epilepsy in children with cerebral palsy. Seizure. 2003 Mar;12(2):110-4. doi: 10.1016/s1059131102002558.
37. Singhi P, Jagirdar S, Khandelwal N, Malhi P. Epilepsy in children with cerebral palsy. J Child Neurol. 2003;18(3):174-9. doi:
38. Fisher RS, Cross JH, French JA, Higurashi N, Hirsch E, Jansen FE, et al. Clasificación operacional de los tipos de crisis por la Liga Internacional contra la Epilepsia: Documento - Posición de la Comisión para Clasificación y Terminología de la ILAE. Epilepsia. 2017;58(4):522-30. doi: 10.1111/epi.13670.
39. World Health Organization. Epilepsy: a public health imperative. 2019. Available from: https://www.who.int/mental_health/neurology/epilepsy/report_2019/en/.
40. Stafstrom CE, Carmant L. Seizures and epilepsy: an overview for neuroscientists. Cold Spring Harb Perspect Med. 2015;5(6):a022426. doi: 10.1101/cshperspect.a022426.
41. Ito S, Nakayama T, Ide S, Ito Y, Oguni H, Goto Y, et al. Aromatic L-amino acid decarboxylase deficiency associated with epilepsy mimicking non-epileptic involuntary movements. Dev Med Child Neurol. 2008;50(11):876-8. doi: 10.1111/j.1469-8749.2008.03094.x.
42. Pons R, Ford B, Chiriboga CA, Clayton PT, Hinton V, Hyland K, et al. Aromatic L-amino acid decarboxylase deficiency: clinical features, treatment, and prognosis. Neurology. 2004;62(7):1058-65. doi: 10.1212/wnl.62.7.1058.
43. Lee W-T. Disorders of monoamine metabolism: inherited disorders frequently misdiagnosed as epilepsy. Epilepsy Seizure. 2010;3(1):147-53. doi: 10.3805/eands.3.147.
44. Swoboda KJ, Saul JP, McKenna CE, Speller NB, Hyland K. Aromatic L-amino acid decarboxylase deficiency: overview of clinical features and outcomes. Ann Neurol. 2003;54 Suppl 6:S49-55. doi: 10.1002/ana.10631.
45. Hsieh HJ, Lin SH, Liu HM. Visualisation of impaired dopamine biosynthesis in a case of aromatic L-amino acid decarboxylase deficiency by co-registered 18F-FDOPA PET and magnetic resonance imaging. Eur J Nucl Med Mol Imaging. 2005;32(4):517. doi: 10.1007/s00259-004-1618-6.
46. Anselm IA, Darras BT. Catecholamine toxicity in aromatic L-amino acid decarboxylase deficiency. Pediatr Neurol. 2006;35(2):142-4. doi: 10.1016/j.pediatrneurol.2006.01.008.
47. Pearson TS, Gilbert L, Opladen T, García-Cazorla A, Mastrangelo M, Leuzzi V, et al. AADC deficiency from infancy to adulthood: Symptoms and developmental outcome in an international cohort of 63 patients. J Inherit Metab Dis. 2020;43(5):1121-30. doi: 10.1002/jimd.12247.
48. Orphanet. Progressive myoclonic epilepsy with dystonia. Available from: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Expert=352596&lng=EN.
49. Dowling JJ, D Gonorazky H, Cohn RD, Campbell C. Treating pediatric neuromuscular disorders: The future is now. Am J Med Genet A. 2018;176(4):804-41. doi: 10.1002/ajmg.a.38418.
50. van Putten M, Hmeljak J, Aartsma-Rus A, Dowling JJ. Moving neuromuscular disorders research forward: from novel models to clinical studies. Dis Model Mech. 2020;13(2):dmm044370. doi: 10.1242/dmm.044370.
51. Mary P, Servais L, Vialle R. Neuromuscular diseases: Diagnosis and management. Orthop Traumatol Surg Res. 2018;104(1S):S89-S95. doi: 10.1016/j.otsr.2017.04.019.
52. Deenen JC, Horlings CG, Verschuuren JJ, Verbeek AL, van Engelen BG. The Epidemiology of Neuromuscular Disorders: A Comprehensive Overview of the Literature. J Neuromuscul Dis. 2015;2(1):73-85.
53. Morrison BM. Neuromuscular Diseases. Semin Neurol. 2016;36(5):409-18. doi: 10.1055/s-0036-1586263.
54. Sáez A, Acha B, Montero-Sánchez A, Rivas E, Escudero LM, Serrano C. Neuromuscular disease classification system. J Biomed Opt. 2013;18(6):066017. doi: 10.1117/1.JBO.18.6.066017.
55. Kaler J, Hussain A, Patel S, Majhi S. Neuromuscular Junction Disorders and Floppy Infant Syndrome: A Comprehensive Review. Cureus. 2020;12(2):e6922. doi: 10.7759/cureus.6922.
56. Adam MP, Ardinger HH, Pagon RA, et al (Editors). GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2020.
57. Schaefer AM, Taylor RW, Turnbull DM, Chinnery PF. The epidemiology of mitochondrial disorders - past, present and future. Biochim Biophys Acta. 2004;1659(2-3):115-20. doi: 10.1016/j.bbabio.2004.09.005.
58. Marecos C, Ng J, Kurian MA. What is new for monoamine neurotransmitter disorders? J Inherit Metab Dis. 2014;37(4):619-26. doi: 10.1007/s10545-014-9697-4.
59. García-Cazorla A, Duarte S, Serrano M, Nascimento A, Ormazabal A, Carrilho I, et al. Mitochondrial diseases mimicking neurotransmitter defects. Mitochondrion. 2008;8(3):273-8. doi: 10.1016/j.mito.2008.05.001.
60. Gropman AL. Neuroimaging in mitochondrial disorders. Neurotherapeutics. 2013;10(2):273-85. doi: 10.1007/s13311-012-0161-6.
61. Opladen T, Cortès-Saladelafont E, Mastrangelo M, Horvath G, Pons R, Lopez-Laso E, et al; International Working Group on Neurotransmitter related disorders (iNTD). The International Working Group on Neurotransmitter related Disorders (iNTD): A worldwide research project focused on primary and secondary neurotransmitter disorders. Mol Genet Metab Rep. 2016;9:61-6. doi: 10.1016/j.ymgmr.2016.09.006.
62. Pousada M, Guillamón N, Hernández-Encuentra E, Muñoz E, Redolar D, Boixadós M, et al. Impact of Caring for a Child with Cerebral Palsy on the Quality of Life of Parents: A Systematic Review of the Literature. J Dev Phys Disabil 2013;25:545-77. doi.org/10.1007/s10882-013-9332-6.
63. Lai ST, Tan WY, Wo MC, Lim KS, Ahmad SB, Tan CT. Burden in caregivers of adults with epilepsy in Asian families. Seizure. 2019;71:132-9. doi: 10.1016/j.seizure.2019.07.008.
64. Landfeldt E, Lindgren P, Bell CF, Guglieri M, Straub V, Lochmüller H, et al. Quantifying the burden of caregiving in Duchenne muscular dystrophy. J Neurol. 2016;263(5):906-15. doi: 10.1007/s00415-016-8080-9.