Capítulo 3
Diagnóstico genético
QUE PROTOCOLO É RECOMENDADO PARA O ESTUDO GENÉTICO DA DISTROFIA MUSCULAR DE DUCHENNE/DISTROFIA MUSCULAR DE BECKER?
P. Gallano Petit
As distrofinopatias são doenças ligadas ao cromossoma X provocadas por mutações no gene da distrofina e incluem a DMD e a DMB. Nos últimos anos, a aplicação de novas tecnologias tem influenciado os testes genéticos de diagnóstico das distrofinopatias.
As deleções ou as duplicações de um ou mais exões são o tipo mutação predominante na patologia molecular do gene da distrofina (~70%). Por conseguinte, o primeiro teste a ser realizado no diagnóstico genético é a análise deste número de cópias exónicas (Fig. 1)1.
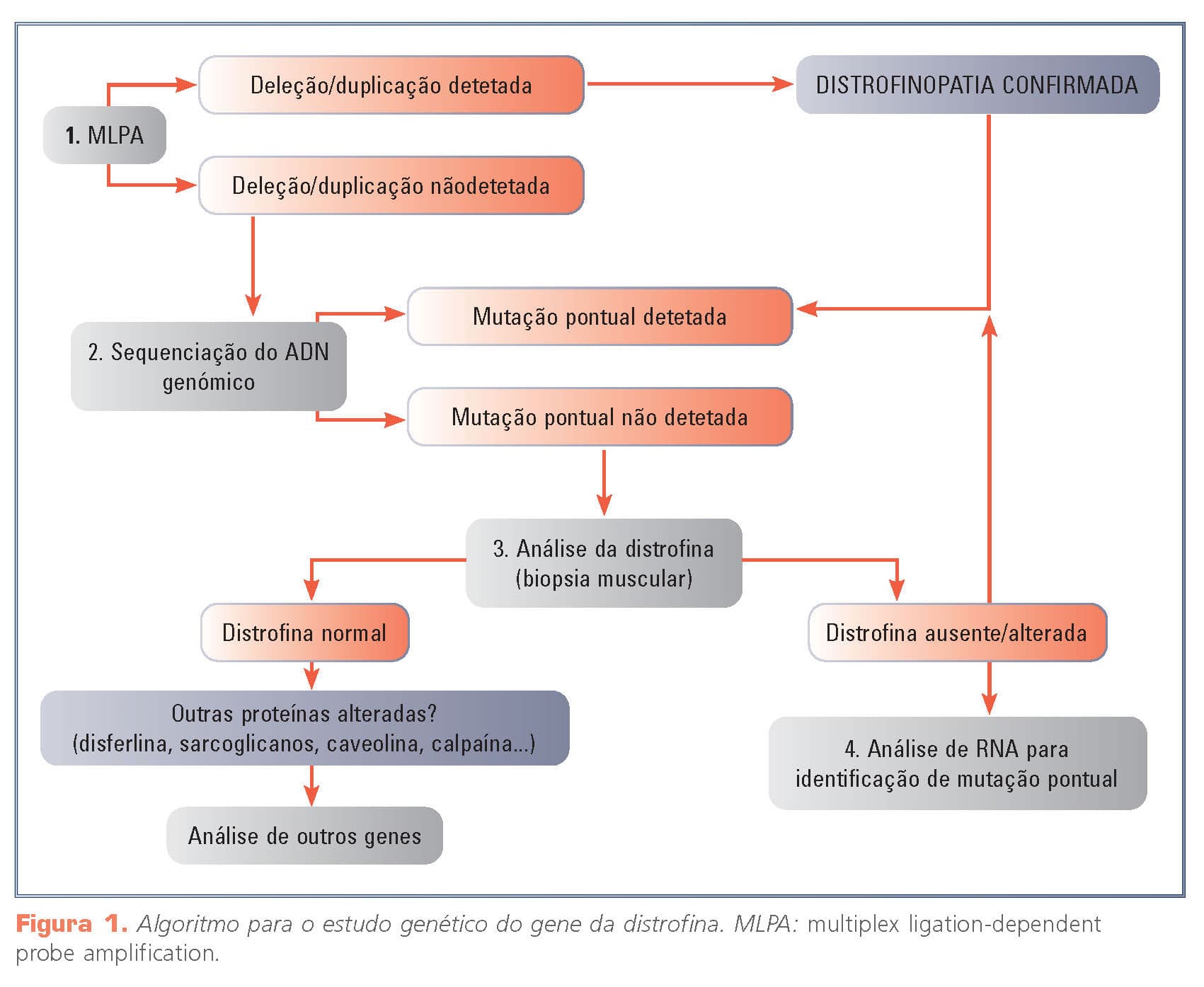
Entre os muitos métodos quantitativos disponíveis, o (multiple ligation probe amplification (MLPA) é atualmente o mais utilizado, devido à sua elevada sensibilidade e baixo custo2. Se for identificada a deleção de um único exão, é necessário confirmar o resultado por um método diferente (por exemplo, através da sequenciação de Sanger do exão em questão) e descartar a possibilidade de que se trata de um falso positivo causado por uma alteração pontual (polimórfica ou patogénica), na sequência de ADN do doente que impede a técnica MLPA3 de funcionar corretamente.
As mutações responsáveis pelos restantes 30% dos doentes consistem em mutações pontuais (nonsense, framehift, pequenas mutações indel). Como as mutações pontuais no gene da distrofina são muito variadas e distribuídas ao longo do gene, as sequências intrónicas flanqueantes e todos os 79 exões devem ser analisados. Isto geralmente é feito através da sequenciação do ADN genómico do doente. Embora seja um método preciso, a sequenciação de Sanger é muito demorada (por causa do grande tamanho do gene da distrofina4), pelo que, neste momento, está a ser substituída pela sequenciação de nova geração (NGS) que é capaz de dar um resultado num período de tempo mais curto5. Se o resultado for negativo, proceder-se-ia, em última instância, à sequenciação do ADNc derivado do RNA obtido da biopsia muscular, o que permitiria identificar a presença de uma mutação pontual.
É IMPORTANTE O ESTUDO GENÉTICO DOS PORTADORES DE DMD?
L. González Quereda
O gene da distrofina está localizado no braço curto do cromossoma X (Xp21)6,7 e é transmitido de forma recessiva, ligado ao cromossoma X. Como os homens têm um único cromossoma X e as mulheres têm dois, se um homem herdar o cromossoma X com a mutação do gene da distrofina, ele adquirirá a doença, enquanto as mulheres serão portadoras, na sua maioria assintomáticas. Uma portadora tem 50% de hipóteses de passar o alelo com a mutação em cada gestação (independentemente do sexo do feto). No entanto, estima-se que cerca de 30% dos casos de distrofinopatias são causados por novas mutações8 , ou seja, mutações que aparecem pela primeira vez na família e não estão presentes nas gerações anteriores. Perante o primeiro caso de DMD numa família, e uma vez identificada a mutação responsável pela patologia no caso índice, é fundamental oferecer um estudo genético à mãe do doente para verificar se ela é portadora da doença. A informação resultante do seu teste genético será essencial para o aconselhamento genético dos outros membros da família. Se a mulher for portadora, ela pode ser geneticamente aconselhada e conhecer o risco de recidiva nos seus descendentes, a probabilidade de retransmitir a doença nas gravidezes futuras e as diferentes opções reprodutivas disponíveis. A informação sobre o estatuto de portadora será também importante para as outras mulheres da família por via materna (irmãs, tias, etc.), que também podem ser portadoras da mutação DMD.
Se o resultado do teste genético revelar que a mulher não é portadora, a probabilidade de transmitir a doença às gerações futuras é consideravelmente menor, apesar de não ser isento de algum risco, dado que existe uma probabilidade de cerca de 7-10% de que a mulher seja portadora de mosaicismo germinativo9. Neste caso, a mutação não será detetada no ADN genómico procedente do sangue periférico, pois a mutação só estará presente em parte das suas células germinativas.
Conhecer o estatuto de portadora oferece também às mulheres a possibilidade de receberem acompanhamento médico, com especial foco para o aspeto cardíaco de modo a detetar precocemente problemas cardíacos que, por vezes, aparecem nas mulheres portadoras10.
QUAL A IMPORTÂNCIA DE UMA INTERPRETAÇÃO ADEQUADA DO ESTUDO GENÉTICO E AS SUAS IMPLICAÇÕES PARA AS OPÇÕES TERAPÊUTICAS?
P. Gallano Petit
O gene da distrofina consiste em 79 exões (regiões codificadoras) ao longo de cerca de 2.200 kb (~2 milhões de nucléotidos) e codifica para a proteína denominada distrofina, cuja isoforma muscular consiste em 3.654 aminoácidos (aa). O código de leitura da distrofina é realizado nos ribossomas, os quais traduzem a mensagem como triplets ou codões graças ao RNA mensageiro que é o ácido nucleico intermediário ou «porta-voz» do ADN genómico. O código genético determina que cada aa que é integrado na cadeia proteica será codificado por três nucléotidos.
O número de nucléotidos que compõem cada exão nem sempre é um múltiplo de três. Muitas vezes, o exão termina com um único nucléotido que é seguido por dois nucléotidos no início do exão seguinte, incorporando assim um novo aa entre os três. Por outro lado, se a sequência exónica terminar com dois nucléotidos, estes serão traduzidos juntamente com o primeiro nucleótido do exão seguinte (Fig. 2)11,12.

Uma das abordagens terapêuticas à DMD é o skipping de exões13. Este tipo de terapia visa restaurar o código de leitura da distrofina dos doentes com DMD que foi interrompido pela presença de uma deleção ou mutação pontual, as quais originaram o aparecimento de um dos três codões de terminação do código genético (UAA, UAG UGA). Assim, por exemplo, o skipping do exão 51 (Fig. 2) restaura a deleção do exão 50, ligando o exão 49 ao exão 52.
Outra abordagem terapêutica é o read-through através do medicamento PTC124, que visa cancelar a leitura dos codões de terminação prematuros produzidos por mutações nonsense, ou seja, aquelas alterações nucleotídicas que criam diretamente um dos três codões de terminação, respeitando simultaneamente a leitura natural do codão de fim de tradução14.
Ao contrário das mutações nonsense, as mutações pontuais do tipo frameshift originam um desfasamento do modelo de leitura da proteína e, em segundo lugar, resultam sempre no aparecimento de um codão de terminação prematuro, a uma certa distância do local da mutação. Este tipo de mutação não pode ser reparada por este tipo de terapia. Assim, em ambas as abordagens terapêuticas é essencial uma definição clara da mutação identificada.
BIBLIOGRAFIA
1. Fratter C, Dalgleish R, Allen SK, Santos R, Abbs S, Tuffery-Giraud S, et al. EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur J Hum Genet. 2020;28(9):1141-59.
2. Lalic T, Vossen RHAM, Coffa J, Schouten JP, Guc-Scekic M, Radivojevic D, et al. Deletion and duplication screening in the DMD gene using MLPA. Eur J Hum Genet. 2005;13:1231-4.
3. Santos R, Goncalves A, Oliveira J, Vieira E, Vieira JP, Evangelista T, et al. New variants, challenges and pitfalls in DMD genotyping: implications in diagnosis, prognosis and therapy. J Hum Genet. 2014;59:454-64.
4. Flanigan KM, von Niederhausern A, Dunn DM, Alder J, Mendell JR, Weiss RB. Rapid direct sequence analysis of the dystrophin gene. Am J Hum Genet. 2003;72:931-9.
5. Gonzalez-Quereda L, Rodriguez MJ, Diaz-Manera J, Alonso-Perez J, Gallardo E, Nascimento A, et al Targeted next-generation sequencing in a large cohort of genetically undiagnosed patients with neuromuscular disorders in Spain. Genes (Basel). 2020;11(5):539.
6. Koenig M, Hoffman EP, Bertelson CJ, Monaco AP, Feener C, Kunkelet LM. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell. 1987;50(3):509-17.
7. Kunkel LM, Monaco AP, Hoffman E, Koenig M, Feener C, Bertelson C. Molecular studies of progressive muscular dystrophy (Duchenne). Enzyme. 1987;38(1-4):72-5.
8. GrimmT, Kress W, Meng G, Müller CR. Risk assessment and genetic counselling in families with Duchenne muscular dystrophy. Acta Myol. 2012;31(3):179-83.
9. Bakker E, van Broeckhoven C, Bonten EJ, van de Vooren MJ, Veenema H, van Hul W, et al. Germline mosaicism and Duchenne muscular dystrophy mutations. Nature. 1987;329(6139): 554-6.
10. Juan-Mateu J, Rodríguez MJ, Nascimento A, Jiménez-Mallebrera C, González-Quereda L, Rivas E, et al. Prognostic value of X-chromosome inactivation in symptomatic female Carriers of dystrophinopathy. Orphanet J Rare Dis. 2012;7:82.
11. Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2(1):90-5.
12. Aartsma-Rus A, van Deutekon JCT, Fokkema IF, van Ommen GJB, den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: An overview of mutation types and paradoxical cases that conform the reading-frame rule. Muscle Nerve. 2006;34:135-44.
13. van Ommen GJ, van Deutekom J, Aartsma-Rus A. The therapeutic potential of antisense exon skipping. Curr Opin Mol Ther. 2008;10(2):140-9.
14. Campbell C, Barohn RJ, Bertini E, Chabrol B, Comi GP, Darras BT, et al. Meta-analyses of ataluren randomized controlled trials in nonsense mutation Duchenne muscular dystrophy. J Comp Eff Res. 2020;9(14):973-84.